Практикум 2. Филогенетическая реконструкция и сравнение деревьев
Для построения выравниваний сначала при помощи команды seqret были получены аминокислотные последовательности цитохромов B выбранных ранее животных (См. практикум 1):
seqret @cyb.list cyb.fasta
Выравнивание было проведено с помощью программы muscle (параметры по умолчанию):
muscle -align cyb.fasta -output cyb-alignment.fasta
На основании данного выравнивания филогенетическое дерево было реконструировано тремя способами: программой FastME, оценивая эволюционные расстояния как p-distance и с помощью модели MtREV, а также программой iqtree.
Реконструирование дерева программой FastME
Для начала выравнивание было переведено в формат “phylip-relaxed”, воспринимаемый программой FastME (см. скрипт). Реконструирование дерева было проведено при помощи команд:
fastme -pp -i cyb.phy -o cyb-p-distance.tre
fastme -pM -i cyb.phy -o cyb-MtRev.tre
Параметры: -p - выбор модели, -i - входной файл, -o - выходной файл
Все полученные деревья были укоренены в ветвь, разделяющую два крупных подотряда Artioctyla: Ruminantia (Жвачные парнокопытные) и Whippomorpha (Кито-бегемотовые. Это устоявшееся разделение, подтвержденное и в современных статьях [1].
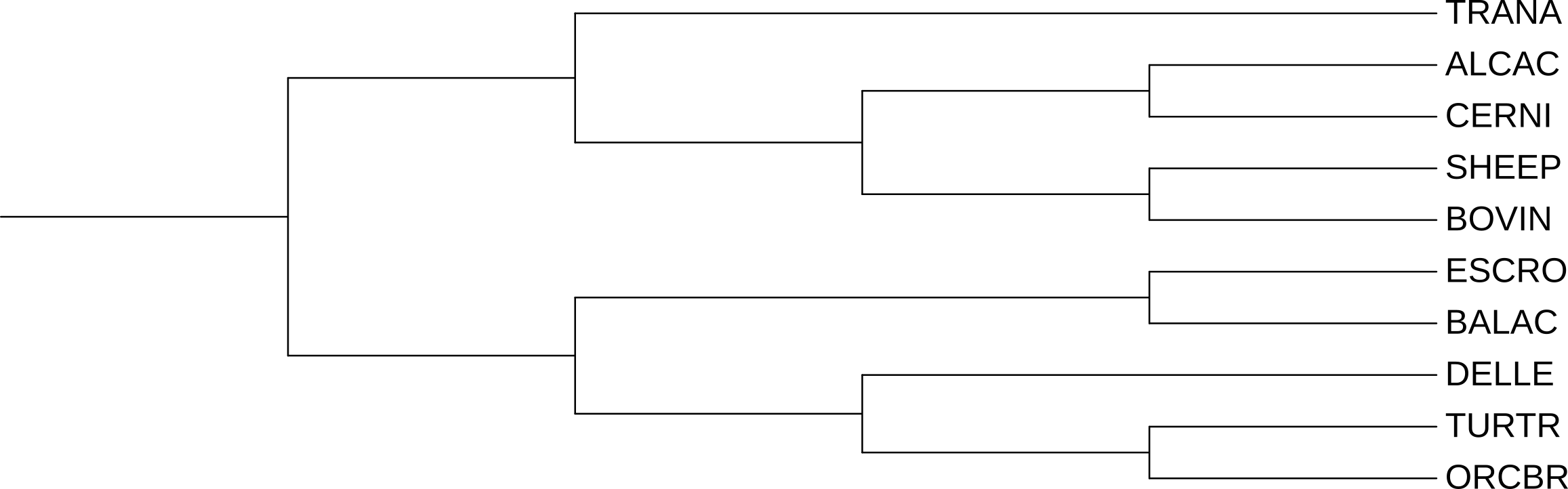
На рисунке 1 представлено дерево, получененное в 1 практикуме на основании общепринятой таксономии, его будем использовать в качестве рефреренса. На рисунке 2 изображено дерево, построенное при оценивании эволюционных расстояний c помощью модели p-distance.
a) p-distance
Строит дерево на основе попарных эволюционных расстояний между последовательностями, которое считается как число отличий / длину последовательности. Соответственно, она не учитывает множественные замены и неодинаковую частоту разных замен.
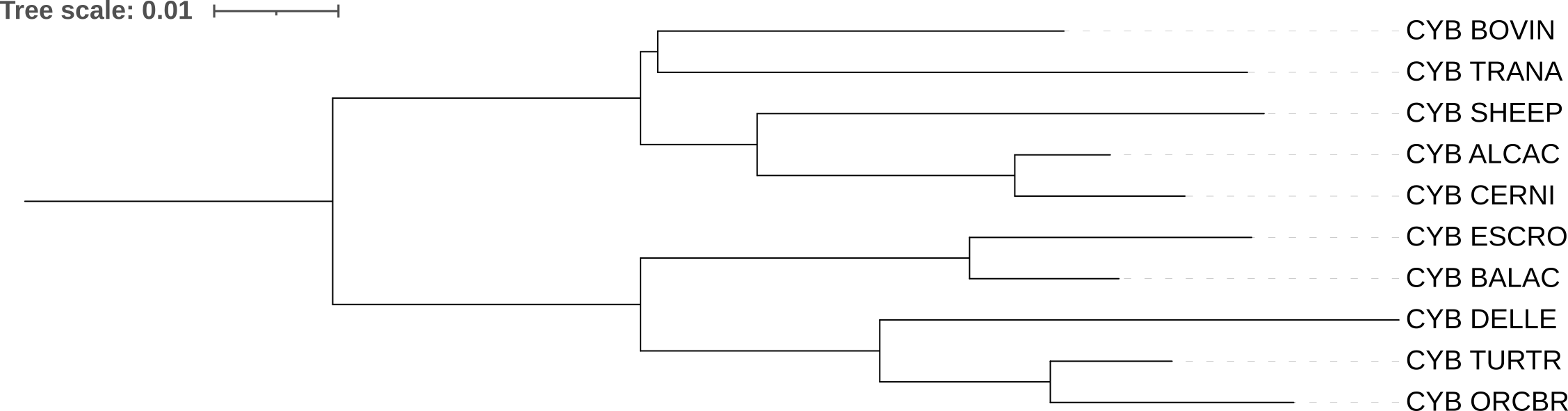
При сравнении полученного дерева с референсом видно, что BOVIN объединился в кладу с TRANA, ветвь теперь не бифурцирует на SHEEP и BOVIN. Клады ALCAC+CERNI и Whippomorpha (Кито-бегемотовые) совпадают. На мой взгляд, это странная реконструкция, так как TRANA (большой оленёк) не относится к Pecora (Настоящие жвачные), представителем которых является BOVIN. Tragulina (оленьковые) отделились от основной линии жвачных очень давно — это самая базальная ветвь среди всех жвачных.
б) MtREV
Модель MtREV оценивает эволюционные расстояние специфично для митохондриальных белков многоклеточных организмов и учитывает особенности замен в них (рис.2)
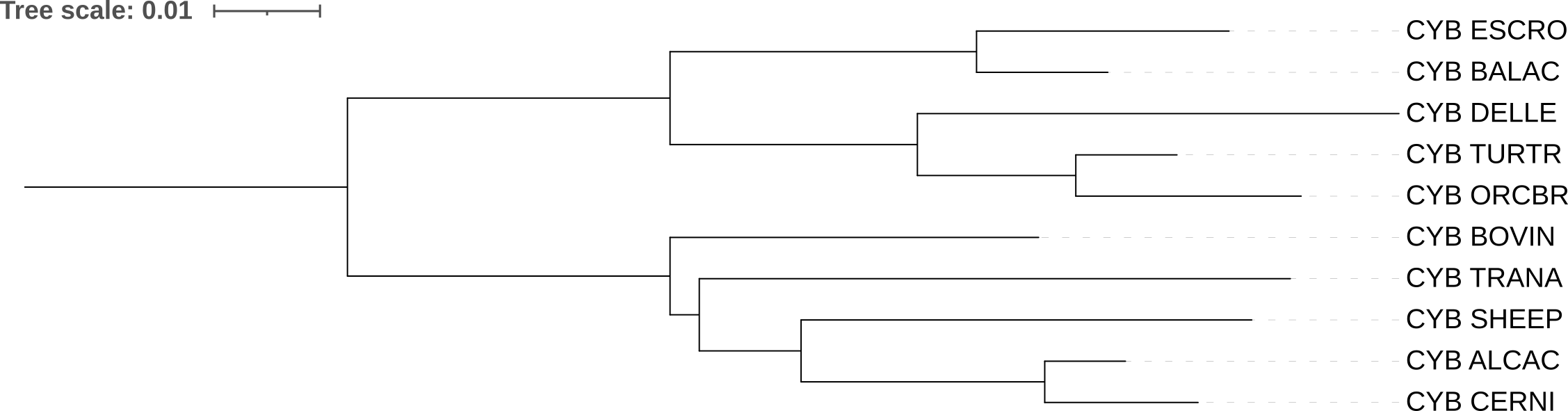
Также на этом дереве BOVIN выделяется в отдельную ветвь в отрыве от жвачных и становится базальной ветвью, в т.ч. для оленьковых, что не является правдой. Клады ALCAC+CERNI и Whippomorpha вновь совпадают с референсом, длины ветвей для этих клад одинаковы в обоих деревьях, построенных программой FastME, что указывает на схожесть алгоритмов построения.
Реконструирование дерева программой IQ-Tree
IQ-TREE строит филогенетическое дерево методом максимального правдоподобия (ML), перебирая множество возможных деревьев и оценивая, какое из них с наибольшей вероятностью объясняет наблюдаемые данные с автоматическим подбором модели эволюции (рис.3)

На этом дереве BOVIN опять объединился в кладу с TRANA, SHEEP остался один. Немного иные длины ветвей, но в остальном это дерево совпадает с референсом (и по топологии полностью повторяет дерево, в котором эволюционные расстояния оценивались как p-distance). Примечательно, что ветвь DELLE (Белуха) демонстрирует наибольшую длину во всех трёх реконструированных деревьях, это наблюдение свидетельствует об ускоренной эволюции митохондриального белка цитохрома B в линии белухи по сравнению с другими исследованными таксонами.
Вывод: филогенетические деревья, построенные программами IQ‑TREE и FastME, в целом соответствуют референсной топологии, однако имеют расхождения в положении отдельных таксонов: оленьковые (Tragulidae) не занимают базальное положение среди жвачных. В то же время основные клады - Cervidae (оленевые) и Whippomorpha (Кито-бегемотовые) воспроизводятся во всех анализах, что указывает на большую надёжность такой филогенетической реконструкции.