| IUPAC definition | Acetylvalylvalyl-4-amino-3-hydroxy-6-methylheptanoylalanyl-4-amino-3-hydroxyl-6-methylheptanoic acid | |||
| Chemical formula | Ac-Val-Val-Sta-Ala-Sta |  |
||
| Molecular formula | C31H57N5O9 | |||
| Molar mass | 643.823 g/mol | |||
| Link to ID in database PubChem | 5481345 |
 |
Intermolecular interactions
The intermolecular interactions of this complex were studied by using the Jmol visualizer.
The following interactions are possible between the chains in the 5HVP structure:
- Hydrogen bonds
- Ionic interaction
- Hydrophobic interaction.
Hydrogen bonds
A hydrogen bond is the electrostatic interaction of a hydrogen atom that is associated with a highly electronegative
element with other atoms. Hydrogen bonds form a hydrogen atom linked to an oxygen or nitrogen atom.
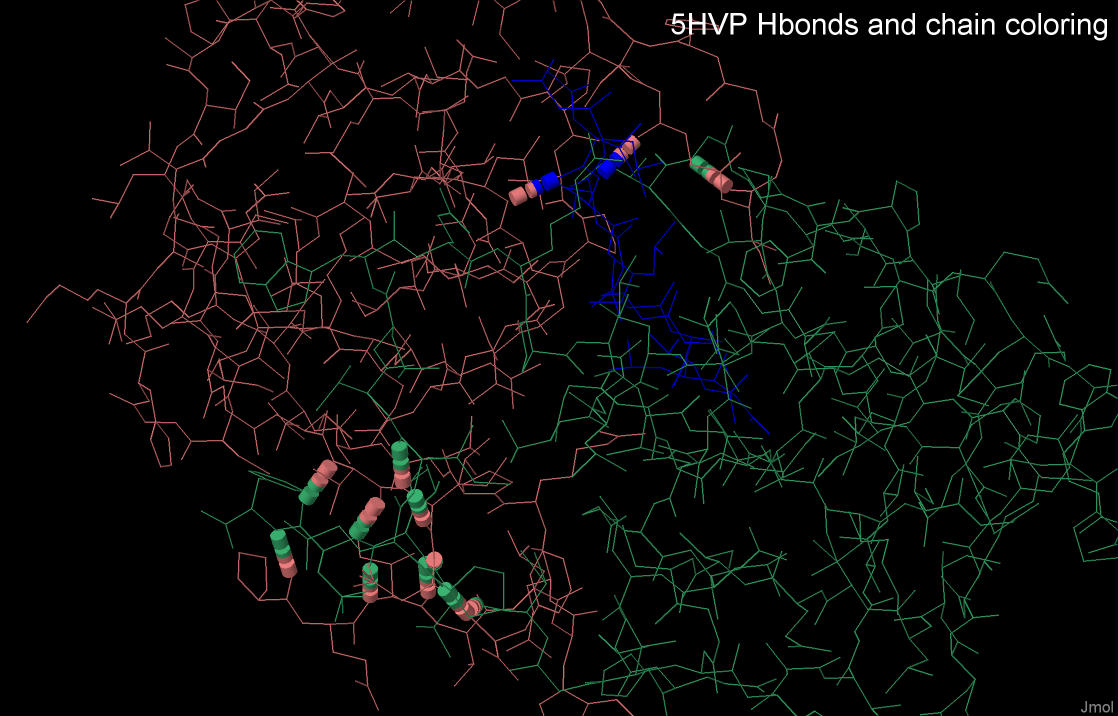
Data on the interchain hydrogen bonds of the 5HVP complex are presented in table 1.
| Total | Between А and В | Between А and С | Between В and С | |
| Quantity | 13 | 11 | 2 | 0 |
| Mean length | 2,91Å | 2,96Å | 2,85Å |
As it can be seen from this table, the majority of interchain hydrogen bonds are formed
between protease subunits, i.e. chains A and B. The ligand molecule forms only two hydrogen bonds with chain A.
In addition, the average length of hydrogen bonds
between the ligand and the protein is slightly less than that between the protease chains.
Hydrophobic interaction in protein structure.
A structure of hydrophobic clusters within the protein was determined by CLuD[4]. It is worth highlighting that there
is only one completed hydrophobic core of the complex, which includes the most of the hydrophobic residues of the protein.
By looking at the protein model, it is clear that the structure of the hydrophobic core passes through both sub-units of the
protein so it can be assumed that the hydrophobic contacts
play the important role in maintaining the tertiary structure of the HIV protease 5HVP.
The packing density of atoms in the hydrophobic core.
The 259th beta-chain residue, tyrosine ([TYR]259:B), was chosen as a hydrophobic residue to study
the packing density of atoms in the hydrophobic core due to the large size of its structure.
To study the hydrophobic core the above-mentioned residue was identified in a script as MyResidue.
Visualization of van der Waals radii of the residue’s environment in the JMol shows that the coating of residue
atoms is occurred by surrounding atoms within the distance of
5 angstroms that shows a close packing of atoms of the hydrophobic core.
An approximate average distance of 3.75 angstroms was established in a model of the complex by manual measurements
of distances between atoms not bound by covalent bonds.
To answer the question whether it is possible to place another atom between nearby, non-covalently bound protein atoms, it is necessary to determine van der Waals radii of atoms. In this work we used the data according to Pauling: carbon-1.85 angstroms, nitrogen-1.54 angstroms, oxygen-1.4 angstroms, sulfur-1.85 angstroms [5]. The water molecule was used as an analyzed atom, because it can be taken as an oxygen atom, neglecting the hydrogen atoms. Taking into account the diameter of oxygen equal to 2.8 angstroms, and the above calculated average distance between non-covalently bonded atoms, 3.75 angstroms, a placement of a water molecule between neighboring atoms is not possible.
Salt bridges
Salt bridge in biochemistry of proteins is comparatively weak ionic bond between positively and negatively charged side chains of protein residues. This type of interactions is considered as electrostatic.
Ionic interactions stabilize the protein structure.
The distance between atoms is no more than 4 Å.
Amino acid residues that are able to form ionic interactions are Asp and Glu (since negatively charged carboxyl groups are carried in the side radical),
Arg, Lys and His (since positively charged atoms are carried in the side radical).
These interactions are subject to the Coulomb law. Free energy of education depends on the environment in which they are localized.
In the inner non-polar region of the protein, this value corresponds to the order - 5 kcal/mol,
and in the subsurface layers of the protein - 10-20 times less.
So, using the Jmol visualizer, an image of salt bridges of the 5HVP protein was obtained.
In addition, using the Jmol visualizer, the number of ionic bonds in the 5HVP complex was calculated:
there are 13 ionic interactions between the amino acids
inside the chains and 2 ionic interactions between the chains A and B.
Author contributions:
The project was completed by a team of first-class first-year bioinformatists,
including Artem Orlov, Vadim Sydlyarchuk and Olga Shigal.
OS studied hidrogen bonds and created the HTML page.
AO researched hydrophobic interactions and the packing density of atoms in the hydrophobic core.
OS and AO jointly set up JSMol applets. VS paid attention to the particularity of this biomolecule,
took the search for information about the
distinctive properties and functions, and also studied the ionic interactions.
References to sources