Практикум 12. Анализ транскриптомов.
В данном практикуме на 15 хромосому были картированы чтения, полученные в результате секвенирования транскриптома.
Использованные команды для вызова программ
Описание использованных команд приведено в таблице 1.
Команда |
Описание |
fastqc chr15.1.fastq |
Анализирует качество чтений |
hisat2 -x ../chr15 -U chr15.1.fastq --no-softclip -S pr12_map.sam |
Строит выравнивание прочтений и референса в sam-формате (опция --no-spliced-alignment убрана, так как сплайсинг следует учитывать) |
samtools view pr12_map.sam -b -o pr12_map.bam |
Переводит sam-файл в бинарный формат |
samtools sort pr12_map.bam pr12_map_sorted |
Сортирует прочтения по координате начала чтения в референсе |
samtools index pr12_map_sorted.bam |
Индексирование бинарного файла |
htseq-count -f bam -s no -i gene_id -m union pr12_map_sorted.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > pr12_reads.count |
Подсчитывает чтения по указанной разметке |
htseq-count -f bam -s no -i gene_id -m intersection-strict pr12_map_str_sorted.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > pr12_reads_str.count |
По сравнению с предыдущей командой меняется опция -m на intersection-strict. |
grep -E -v '0$' pr12_reads.count |
Вытаскивает строки с попаданием чтений на гены |
Анализ качества чтений
Был получен html-отчёт анализа качества чтений. Основные данные о качестве приведены на рисунке 1.
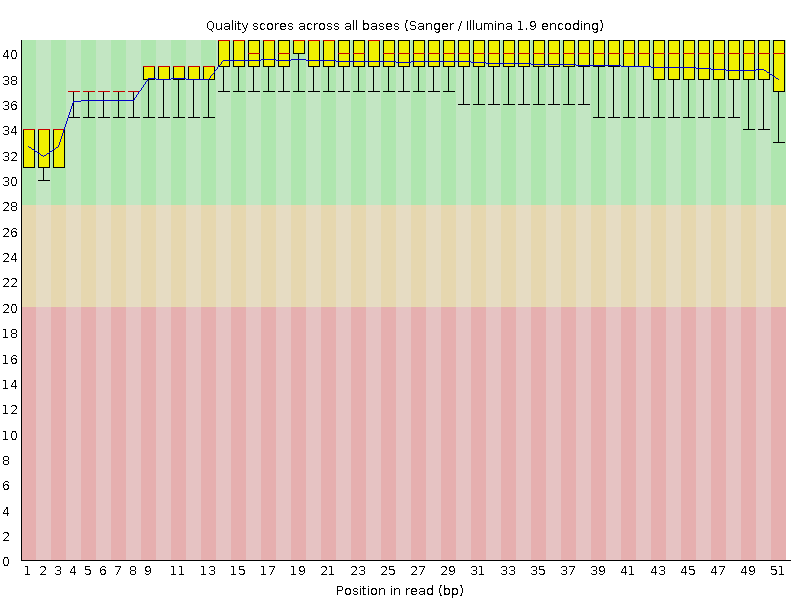
Рисунок 1. Качество чтений оказалось хорошим, так как чтения обладают качеством более 28.
Картирование чтений
Чтения были картированы на 15 хромосому (аналогично предыдущему практикуму) с учётом сплайсинга. Вывод программы:
9891 reads; of these:
9891 (100.00%) were unpaired; of these:
44 (0.44%) aligned 0 times
9847 (99.56%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.56% overall alignment rate
Картирование вышло хорошим, только 0.44% процента от числа чтений не попали в выравнивание. Затем после обработки программами samtools чтения были визуализированы в IGV (рисунок 2).
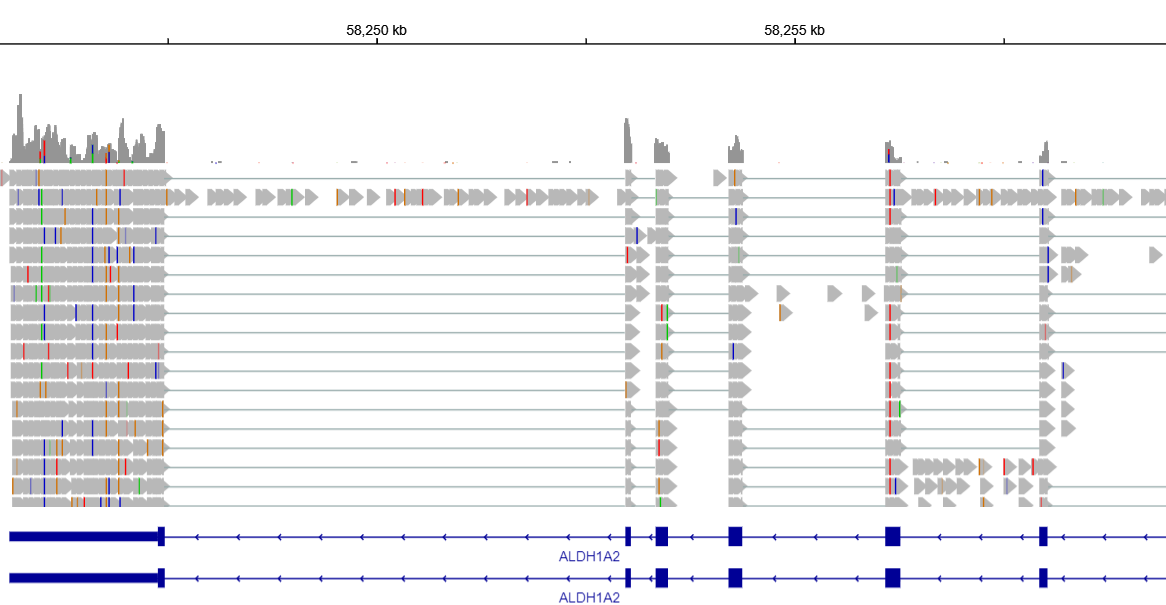
Рисунок 2. В IGV видно, что в транскриптомных чтениях заметны места интронов, разделяющих чтения. Подобное отсутствует при геномных прочтениях.
Подсчет чтений
Были подсчитаны чтения по gtf-разметке. Затем получился результат grep:
ENSG00000259285.1 12 ENSG00000259477.1 14 __no_feature 2505 __ambiguous 326 __not_aligned 44
2505 чтений не попали в гены, для 326 возникла спорная ситуация (например, попадание в 2 гена одновременно), а 44 чтения не были выровнены. Первый ген - длинная некодирующая РНК. Другой ген - это процессированный псевдоген калпонина-2, белка, связанного с клеточной пролиферацией, подвижностью и адгезией.
Всё вышеописанное в практикуме было также сделано для реплики (chr15.2.fastq). Результат:
ENSG00000128918.10 5408 ENSG00000259285.1 4 __no_feature 2267 __ambiguous 159 __not_aligned 35
Ген с 5408 попаданиями чтений кодирует альдегиддегидрогеназу, фермент, окисляющий альдегиды до кислот. По сравнению с репликой, чтений, попавших в гены, стало гораздо больше.
Для chr15.2.fastq c полностью попавшими чтениями в гены (опция -m intersection-strict):
ENSG00000128918.10 5378 ENSG00000259285.1 5 __no_feature 2357 __ambiguous 98 __not_aligned 35
В целом число попаданий чтений в ген уменьшилось, но изменение опции разрешило более трети спорных ситуаций.