1. Сравнение алгоритмов множественных выравниваний
Для выравнивания доменов c PF00060 из seed воспользовалась кодом прграммы MACHO Виталия Гагарочкина. Программа выдала следующие результаты сравнения выравниваний Muscle c Mafft и Muscle с Tcoffee (гиперссылка на проект Jalview):
| Блок | Muscle.fa и Mafft.fa |
| Совпадение 1 | (1,8)=(1,8) |
| Несовпадение 1 | (9,48), (9,52) |
| Совпадение 2 | (49,52)=(53,56) |
| Несовпадение 2 | (53,110), (57,116) |
| Совпадение 3 | (111,125)=(117,131) |
| Несовпадение 3 | (126,130), (132,136) |
| Совпадение 4 | (131,160)=(137,166) |
| Несовпадение 4 | (160,212), (167,) |
| Совпадение 5 | (213,213)=(225,225) |
| Несовпадение 5 | (214,231), (224,244) |
| Совпадение 6 | (232,233)=(245,246) |
| Несовпадение 6 | (234,306), (247,324) |
| Совпадение 7 | (307,307)=(323,323) |
| Несовпадение 7 | (308,316), (324,332) |
| Совпадение 8 | (317,317)=(333,333) |
| Несовпадение 8 | (318,318), (334,334) |
| Совпадение 9 | (319,350)=(335,366) |
| Несовпадение 9 | (351,397), (367,422) |
| Совпадение 10 | (398,403)=(423,428) |
| Несовпадение 10 | (404,405), (429,430) |
| Совпадение 11 | (406,408)=(431,433) |
| Блок | Muscle.fa и Tcoffee.fa |
| Совпадение 1 | (1,8)=(1,8) |
| Несовпадение 1 | (9,14), (9,14) |
| Совпадение 2 | (15,30)=(15,30) |
| Несовпадение 2 | (31,50), (31,50) |
| Совпадение 3 | (51,56)=(51,56) |
| Несовпадение 3 | (57,104), (57,106) |
| Совпадение 4 | (105,105)=(107,107) |
| Несовпадение 4 | (106,107), (108,109) |
| Совпадение 5 | (108,125)=(110,127) |
| Несовпадение 5 | (126,130), (128,132) |
| Совпадение 6 | (131,166)=(133,168) |
| Несовпадение 6 | (167,187), (169,189) |
| Совпадение 7 | (188,188)=(190,190) |
| Несовпадение 7 | (189,232), (191,255) |
| Совпадение 8 | (233,236)=(256,259) |
| Несовпадение 8 | (237,247), (260,238?) |
| Совпадение 9 | (248,249)=(239,240) |
| Несовпадение 9 | (250,311), (241,336) |
| Совпадение 10 | (312,314)=(337,339) |
| Несовпадение 10 | (315,316), (340,341) |
| Совпадение 11 | (317,317)=(342,342) |
| Несовпадение 11 | (318,318), (343,343) |
| Совпадение 12 | (319,359)=(344,384) |
| Несовпадение 12 | (360,391), (385,429) |
| Совпадение 13 | (392,403)=(430,441) |
| Несовпадение 13 | (404,406), (442,444) |
| Совпадение 14 | (407,408)=(445,446) |
Таким образом вы нашли больше совпадающих блоков для Muscle с Tcoffee, что может указывать на большую схожесть двух этих алгоритмов. Программа MACHO также подсчитала, что у данной пары выравниваний больше процент совпадающих колонок (37,01% для Muscle и 33,86% для Tcoffee), нежели у пары Muscle И Mafft (25,25% и 23,79% соответсвенно). В то же время для пары Muscle Mafft характерны длинные малочисленные общие блоки, а для Muscle tcoffee ¯ менее протяженные блоки, но их больше.
2. Сравнение PDBeFold Multiple Alignment и MSAprobes
Я нашла три структуры белка из 26 известных для моего домена fn1 из практикума 11, которым соотвествуют разные белки (разные доменные архитектуры). Это записи с PDB id: 1fbr, 1tpm и 8os5, которым соответсвуют белки из UniProt c AC P02751, P00748, P00750 соответсвенно. Я загрузила их в PDBeFold Multiple Alignment (для 8os5 цепь А), загрузила их выравнивания в fasta формате в jalview, а также выровняла те же pdb id алгоритмом MSAprobes (гиперссылка на проект с двумя выравниваниями). Результаты выравниваний и их сравнения представлены ниже.
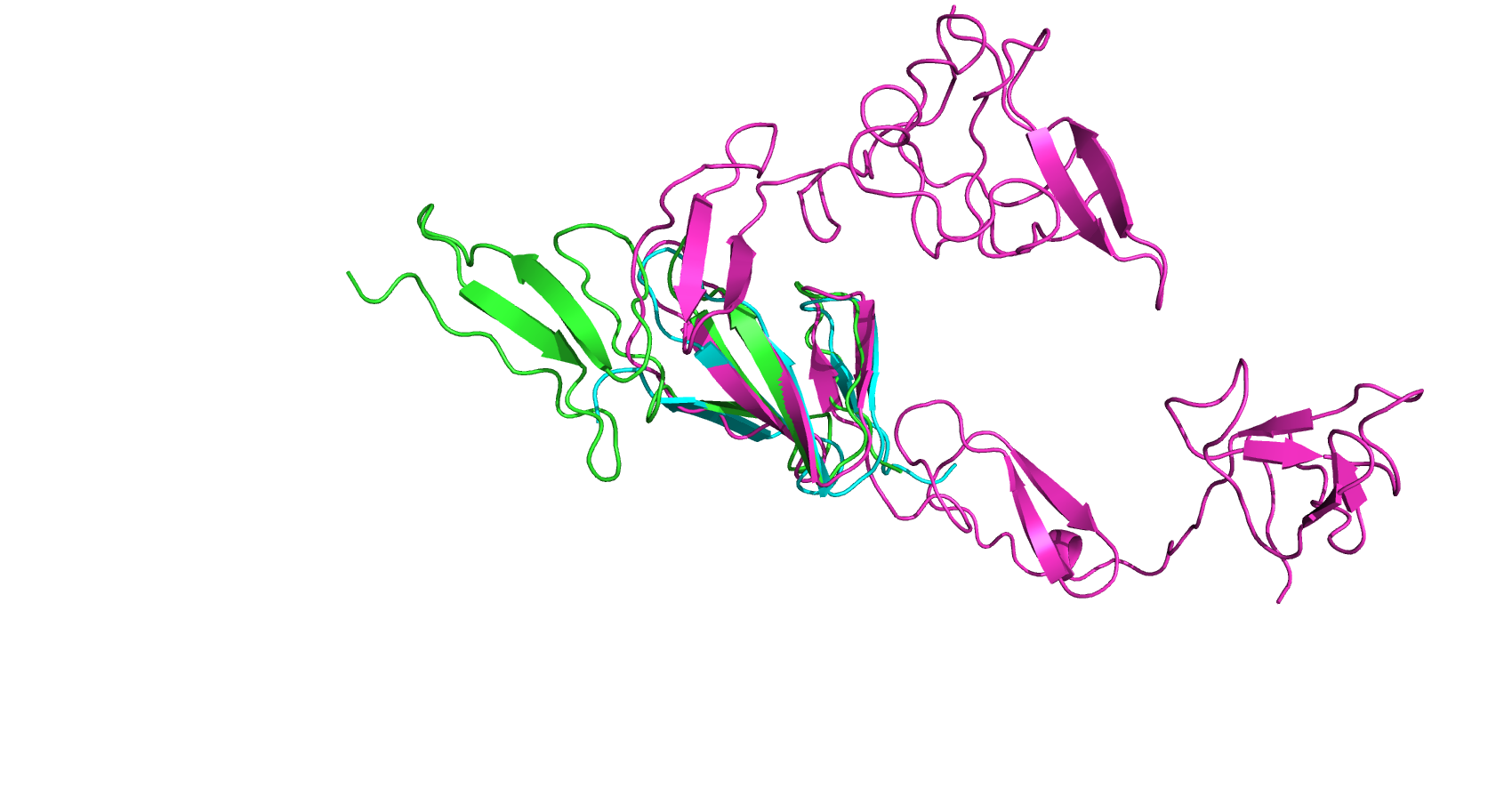


По рис 1. видно, что структурное выравнивание выровняла не весь домен целиком, а только его часть, состоящую из трёх цепей бета-листа, в то время как ещё две цепи второго бета листа остались не выровняны. Вероятно это связано с неструктурированными участками между этими листами, что затрудняет наложение этих структур для выравнивания.
В выравнивании из рис 2, в сравнение с выравниванием из рис 3 (MSAprobes), консервативных колонок меньше, выровнялся не весь домен целиком (участок короче 40 ак, которые составляют домен). В то время как в выравнивании MSA блок выравнивания покрывает домен почти целиком, что видно по вторичной структуре, приведенной ниже. У этих двух выравниваний блок сходства не приходиится на наш домен, во всех колонках, кроме первых 58 (1,58)=(1,58), белки выровняны по-разному, в тч на участке с последовательностью домена.
Таким образом можно заключить, что хоть структурное выравнивание является более наглядным, а также способно отражать структурную схожесть белков, мало связанную с консервативностью последовательности ак, алгоритмы множественного выравнивания MSA справляются с задачей множественного выравнивания лучше. В данном случае выравнивание MSA достаточно точно отражало сходство между доменными архитектурами выбранных белков, в сравнение со структурным.
3. Описание алгоритма работы MSAprobes
В основе работы данного алгоритма лежит совмещение принципа парных скрытых Марковских моделей pair-HMM и статистические суммы свободной энергии (СССЭ, partition function). Простым языком, HMM-профиль, в отличие от классических матриц замен (таких как BLOSSUM), определяет вероятность аминокислотной замены не в целом, а на конкретной позиции в белковой последовательности. Например, замены в активном центер будут штрафоваться больше, чем на неупорядоченном участке. СССЭ реализуется через алгоритм Forward-Backward. То есть сначала алгоритм находит всевозможные выравнивания, не отбрасывая все, кроме лучшего, как это делают классические алгоритмы (например Нидлмана-Вунша), а собирая все эти вероятности в полную вероятность, так называемую статистическую сумму (Z), по аналогии со статической механикой, и энергию каждого такого "состояния" (выравнивания) мы можем нормировать на Z, и при финальном прогрессивном выравнивании зафиксировать самую выгодную позицию. В итоге мы получаем матрицу вероятности для каждой пары белков. Для выставления подходящих штрафов за гэпы, от которых сильно зависит точность алгоритма, используется partial function: какую долю всевозможных выравниваний составляют выравнивания с таким сопоставлением ак из 1 и второй последовательности. По сути мы получаем ещё одну матрицу вероятностей, после чего алгоритм сопоставляет и усредняет их, мы получаем матрицу апостериорной вероятности (вектор точности), который определяет вероятность предложенного соответсвия.
После чего алгоритм вводит дополнительные веса для уточнения позиции выравнивания на древе и строит филогенетичсекое древо по методу UPGMA (прогрессивный алгоритм), в соответсвие с которым группы белков будут объединяться. Здесь тоже есть несколько этапов. Сначала на основе "сглаженных" матриц строят матрицу расстояний, а потом уже древо белков.
Сайт, посвященный выравниванию. На основании статьи Liu, Y., Schmidt, B., & Maskell, D. L. (2010). MSAProbs: multiple sequence alignment based on pair hidden Markov models and partition function posterior probabilities. Bioinformatics, 26(16), 1958–1964.