EMBOSS
Упражнение 1.
В данном упражнении было необходимо несколько файлов в формате fasta собрать в единый файл с помощью
команды seqret. Для этого я выбрал несколько файлов с белковыми последовательностями в формате
fasta: O14746.fasta, O75390.fasta,
P11142.fasta, P24928.fasta. Все эти последовательности
использовались в предыдущем практикуме. Затем с помощью команды
ls > list скопировал имена всех файлов с последовательностями в файл list,
после чего удалил из него название самого файла. Затем командой seqret @list ex_1.fasta сохранил
все файлы в одном - ex_1.fasta.
Упражнение 2.
В данном упражнении было необходимо один файл в формате fasta с несколькими последовательностями
разделить на отдельные fasta файлы с помощью команды seqretsplit. Затем командой
seqretsplit ex_1.fasta разделил файл, полученный в предыдущем упражнении, на четыре отдельных файла:
tert_human.fasta, cisy_human.fasta,
hsp7c_human.fasta, rpb1_human.fasta.
Упражнение 3.
В данном упражнении было необходимо из файла с хромосомой в формате .gb вырезать три кодирующих
последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле.
ДляДля этого я выбрал последовательность хромосомы Brucella ovis. В файле
list_1 я поместил координаты трёх генов, после чего команда seqret @list_1 ex_3.fasta
вырезала эти три последовательности и поместила их в файл ex_3.fasta.
Упражнение 4.
В данном упражнении было необходимо транслировать кодирующие последовательности, лежащие в одном
fasta файле, в аминокислотные, используя указанную таблицу генетического кода, с помощью команды
transeq. Для этого я взял файл с последовательностями, полученный в предыдущем Упражнении, и
командой transeq ex_3.fasta ex_4.fasta -frame 1 транслировал их в аминокислотную последовательность.
Результат - файл ex_4.fasta.
Упражнение 5.
В данном упражнении было необходимо транслировать данную нуклеотидную последовательность в шести
рамках. Для этого я использовал всё тот же файд, полученный в Упражнении 3, и командой
transeq ex_3.fasta ex_5.fasta -frame 6 транслировал каждую из трёх последовательностей по шести
рамкам.
Сравнение аннотаций генов белков в одной хромосоме бактерии или археи с трансляциями длинных
открытых рамок считывания.
Задание 1.
В данном задании было необходимо получить таблицу длинных открытых рамок считывания и трансляции
этих рамок. Для работы я выбрал бактерию, с которой работал в первом семестре - Sulfurihydrogenibium
azorense, - водородокисляющую термофильную бактерию. У неё только одна хромосома.
Файл в формате GenBank, последовательность
генома в fasta-формате.
Командой getorf S_azorense.fasta orf.fasta -table 11 -minsize 180 -find 0 -circular я нашёл
все открытые рамки считывания длиной не менее 180 нуклеотидов (опция -minsize 180), при транслировании
которых использовалась бактериальная таблица генетического кода (опция -table 11) и транслирование
которых проходило от старт- до стоп-кодона. Опция -circulare показывает, что ДНК кольцевая.
Затем командой infoseq orf.fasta -outfile orf.xls -only -name -length -description получил
таблицу с минимальной информацией о рамках (номер, длина, описание). После этого подправил её с
помощью MS Excel (функция ЕСЛИ), и получил таблицу в чистовом варианте.
Файл с транслированными открытими рамками считывания, таблица
со списком найденных рамок.
Задание 2.
Список аннотированных белков бактерии я получил, перейдя со
страницы
генома бактерии по ссылке protein count.
Данную таблицу я скачал на компьютер, удалил лишние столбцы, и получил искомый
результат. Белковые последовательности в виде архива с файлом
получил, перейдя со страницы генома по ссылке Download sequences in FASTA format for protein, после
чего переименовал файл в S_azorense_prots.fasta для удобства.
Задание 3.
При сравнении таблиц с открытими рамками и аннотированными белками можно заметить, что количество
белков (1667) сильно меньше количества рамок (5065), то есть, далеко не каждая рамка соответствует белку.
Для выполнения этого задания я объеденил две таблицы, полученные в Заданиях 1 и 2, и получил
итоговую таблицу. Ниже приведены примеры расхождения в таблицах.
Несовпадение длин транслята рамки считывания и белка. Такое несовпадение может быть связано с тем,
что рамки начинаются только с кодона AТG, а для бактерий характерно существование других старт-кодонов:
GTG, CTG, TTG, ATT. Пример:

Несколько рамок считывания внутри одного белка. Команда getorf могла посчитать началом рамки
считывания любые три нуклеотида, складывающиеся в "кодон" ATG, так что ничего удивительного нет в том, что
внутри одного белка таких "кодонов" нашлось сразу несколько, благо, белок длинный.
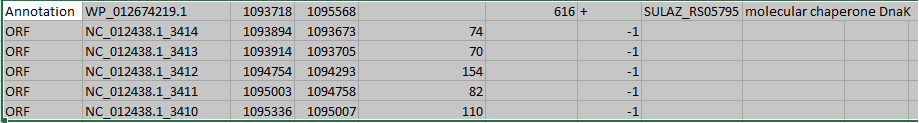 Аналогичный пример, в котором, к тому же, рамки с разных цепей ДНК.
Аналогичный пример, в котором, к тому же, рамки с разных цепей ДНК.
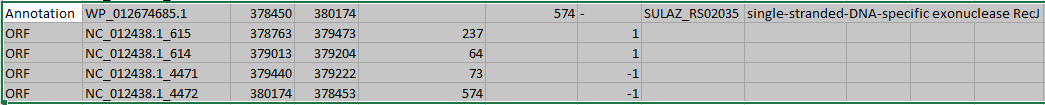
Совпадение длин транслята рамки и белка. Начало обеих последовательностей одинаково, но последовательность
рамки заканчивается на 3 нуклеотида раньше, чем белковая. Это может быть связано с тем, что getorf
не учитывает стоп-кодоны в конце открытой рамки при указании её координат.

Сдвиг рамки на один кодон относительно белка. Видно, что рамка начинается на три нуклеотида раньше
последовательности, кодирующей белок, и на три же нуклеотида раньше заканчивается. Это может быть связано
с тем, что прямо перед старт-кодоном в гене белак стоит кодон ATG в рамке, а стоп-кодон в последовательности
рамки, как уже было сказано, не учитывается.

Для длинного белка не нашлось подходящей рамки (все вероятно подходящие рамки сильно короче). Видимо,
по всей длине белка нашёлся лишь один кодон ATG или нуклеотиды в данном порядке, чтобы началась рамка,
лишь краешком пересекающаяся с последовательностью, кодирующей белок. Этот феномен можно объяснить тем,
что метионин (кодируемый кодоном ATG) - довольно редкая аминокислота.

Пересечение антипараллельных рамок на более чем 150 нуклеотидов. Видим, что в данном примере достаточно
длинная рамка на обратной цепи, фактически, лежит в пределах границ рамки прямой цепи, в которой, к тому же,
закодирован белок.

Для белка не нашлось подходящей рамки (все вероятно подходящие рамки сильно длиннее). Возможно, это
связано с тем, что белок начинается с какого-либо другого старт-кодона (не ATG), а внутри кодирующей его
последовательности ДНК только один раз нашлись нуклеотиды, складывающиеся в кодон ATG, а стоп-кодон
для рамки наступил не скоро. Такое различие в номерах концевых нуклеотидов между белком и рамкой, видимо,
связано с тем, что рамка считывания белка сдвинута на 1-2 нуклеотида относительно рамки, найденной командой
getorf.

Ссылка на главную страницу
© Головачев Ярослав