A- и В-формы ДНК. Структура РНК
Модели форм ДНК
С помощью инструментов пакета 3DNA, включающего программы для анализа и моделирования структурнуклеиновых кислот и работающего под LINUX, были построены модели структур A-, B- и Z-формы ДНК. С помощью программы fiber из пакета 3DNA комплементарно сопоставлены две цепи нуклеиновых кислот, состоящие из пятикратного повторения последовательности "gatc", с образованием дуплекса. Результат данной работы визуализирован и представлен на трех апплетах ниже, при запуске которых Вы сможете изучить формы подробнее, а по ссылкам доступны файлы в формате pdb.
| А-форма ДНК | B-форма ДНК | Z-форма ДНК |
Сравнение структур ДНК
 |
Для выполнения данного задания в программе JMol была визуализирована B-форма ДНК и опредлены малая и большая бороздки в ней. Затем с помощью программы MarvinSketch для 12 по счету цитозина отмечались атомы, обращенные в сторону большой бороздки - синим, а в сторону малой бороздки - красным. На Рис. 1 представлен результат этой работы, а ниже непосредственно указаны номера атомов, характеризующиеся тем или иным положением в ДНК.
В сторону большой бороздки обращены атомы c12.c6, c12.c5, c12.c4, c12.n4
В сторону малой бороздки обращены атому c12.n1, c12.c2, c12.o2, c12.n3
Используя программу JMol, в Таблицу 1 были сведены некоторая информация о A-, B- и Z-формах ДНК. Для измерения ширины большой и малой бороздок выделялись атомы фосфора в структуре ДНК, а затем измерялись расстояния между атомами комплементарных цепей, находящихся на минимальном расстоянии.
| A-форма | B-форма | *Z-форма | |
| Тип спирали (правая или левая) | Правая | Правая | Левая |
| Шаг спирали (Å) | 28.03 | 33.75 | 43.50 |
| Число оснований на виток | 11 | 10 | 12 |
| Ширина большой бороздки (Å) | 16.81 ([DC]44:B.P - [DT]7:A.P) | 17.21 ([DG]29:B.P - [DG]17:A.P) | 18.30 ([DC]6:A.P - [DC]32:B.P) |
| Ширина малой бороздки (Å) | 7.98 ([DA]6:A.P - [DG]37:B.P) | 11.69 ([DT]15:A.P - [DA]38:B.P) | 8.68 ([DG]31:B.P - [DG]15:A.P) |
Параметры структур нуклеиновых кислот
Далее с помощью программ find_pair и analyze из пакета 3DNA были сравнены некоторые параметры в структурах ДНК (PDB = 1dc1) и тРНК (PDB = 2fmt), находящихся в комплексе с некоторыми белками. В частности, анализировались торсионные углы (вокруг какой-либо связи) - это двугранные углы, определяющие взаимный поворот частей молекулы, находящихся по разные стороны от этой связи [1].
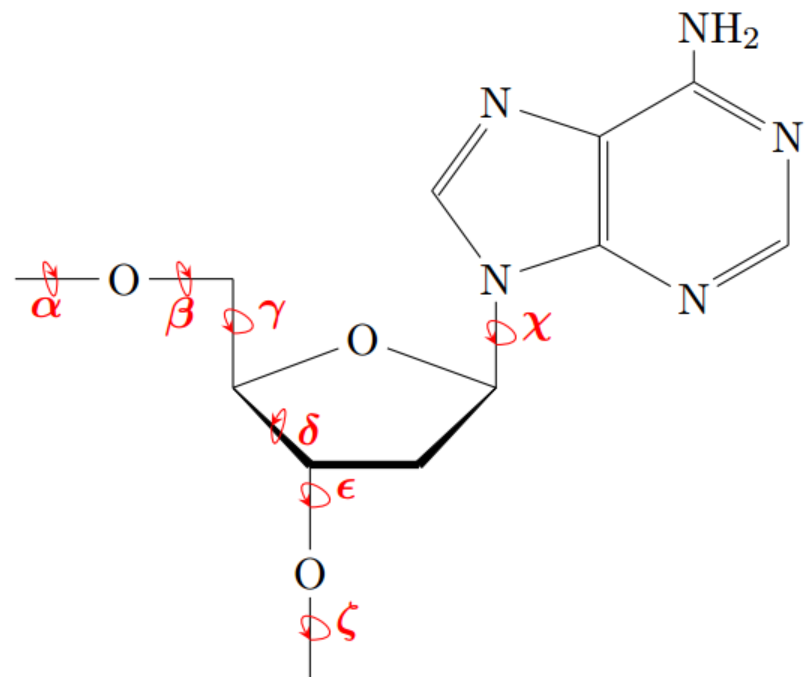 |
Данные, полученные при анализе, были усреднены при помощи стандартных функций (СУММ, СРЗНАЧ) программы Excel, а затем собраны в Таблицу 2, в которой помимо данных для ДНК (PDB = 1dc1) и тРНК (PDB = 2fmt; комплекс формилтрансферазы с двумя(!) молекулами тРНК), Помимо этого в Таблице 2. 1 приведены данные для A- и B-форм ДНК, чтобы определить, на какую из форм похожа структура тРНК. Для проведенения анализа выбран участок тРНК, образующий стебель, как будет показано ниже, а именно с 49 по 53 и с 59 по 63 нуклеотиды. Как видно из таблицы, по значениям углов данная тРНК больше похожа на A-форму ДНК.
Также с помощью программы Excel были определены самые "деформированные" нуклеотиды, то есть с наиболее отклоняющимся значением какого-либо из торсионных углов. Такими являются для тРНК:
Отклонение в большую сторону - 49G, 33U, 32C, 48C, 9G
Отклонение в меньшую сторону - 19G, 13C, 25C, 7G
В одной цепи - 8G, 5C, 2A
В другой цепи - 5G, 6C, 8C, 9A (по двум значениям углов)
Структура тРНК
Для нахождения нуклеотидов, образующих стебли, с помощью программы из пакета 3DNA (find_pair -t 2fmt.pdb stdout | analyze) был получен файл 2fmt_old.out, в котором содержались данные, из которых выбирались пары нуклеотидов, обуславливающие третичную структуру тРНК. На Рис. 3 эти участки визуализированы с помощью JMol, где оранжевым цветом отображен участок с 1 по 6 пары, желтым - с 7 по 8 пары, синим - с 15 по 19, зеленым - с 22 по 25.
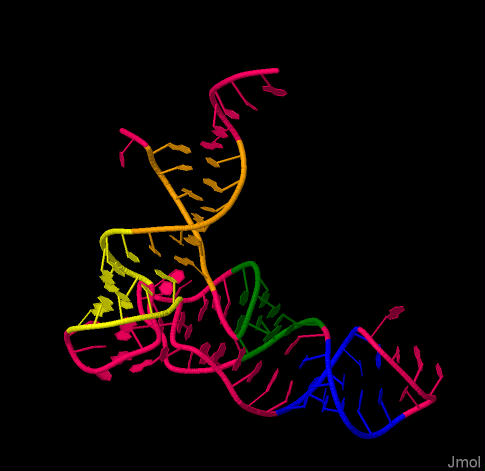 |
|
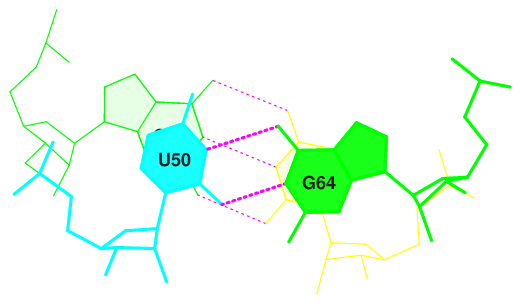
Помимо этого, в структуре тРНК существуют дополнительные водородные связи, стабилизирующие третичную структуру.
В строении РНК часто встречаются неканонические пары оснований. Так, для данной тРНК можно заметить следующие пары.
Стэкинг взаимодействия
Для данного задания была использована та же молекула тРНК, что изучалась выше (PDB = 2fmt). Из файла, полученного путем использования программ find_pair и analyze из пакета 3DNA, 2fmt_old.out были выбраны пары нуклеотидов с наибольшей и наименьшей площадями перекрывания, соответственно.
Далее, используя следующие команды, были получены изображения данных нуклеотидных пар в формате .ps, конвертируемые с помощью специального сайта: ex_str -* stacking.pdb step*.pdb и stack2img -cdolt step*.pdb step*.ps, где вместо * нужно поставить номер анализируемой пары. На Рис. 4 - 6 представлены пары с наибольшей площадью перекрывания. Для наглядности приведены результаты визуализации взаимного расположения этих пар в программе JMol, рассмотренного с разных ракурсов.
 |
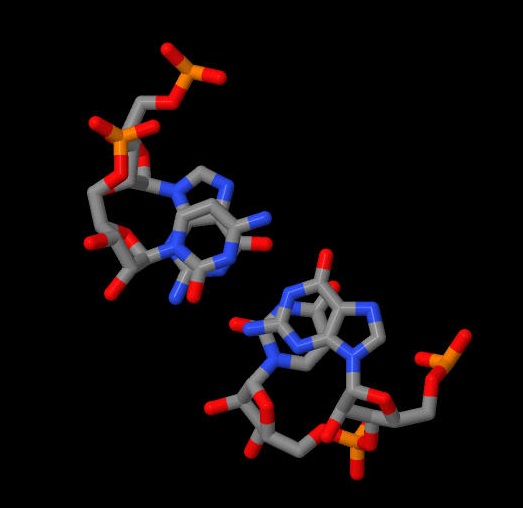 |
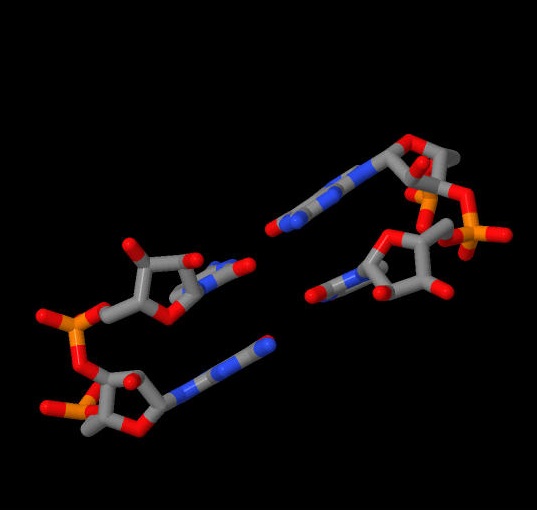 |
На Рис. 7 - 9 представлены пары с наименьшей площадью перекрывания. Для наглядности приведены результаты визуализации взаимного расположения этих пар в программе JMol, рассмотренного с разных ракурсов.
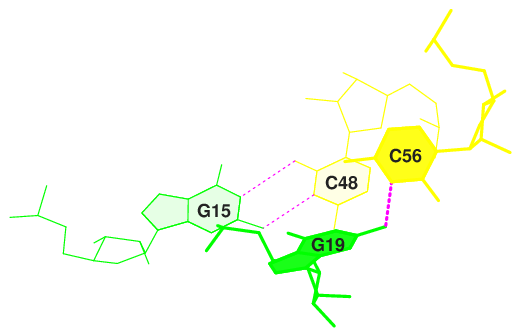 |
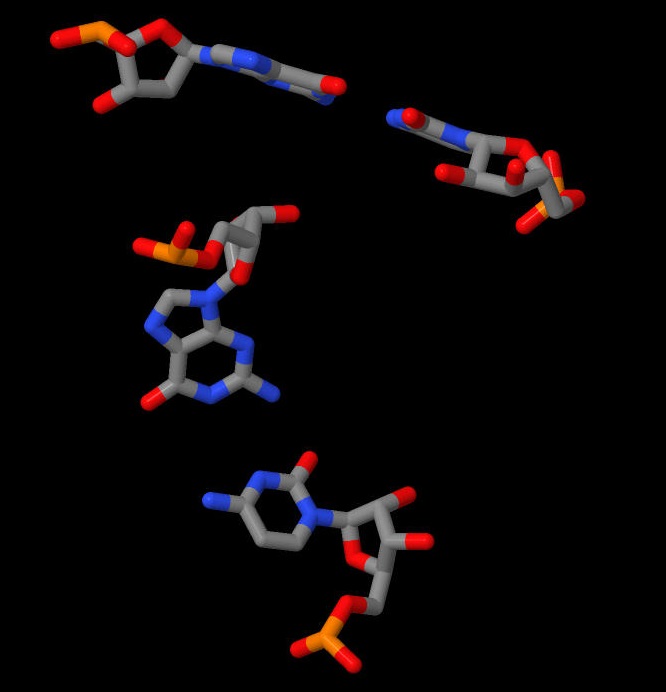 |
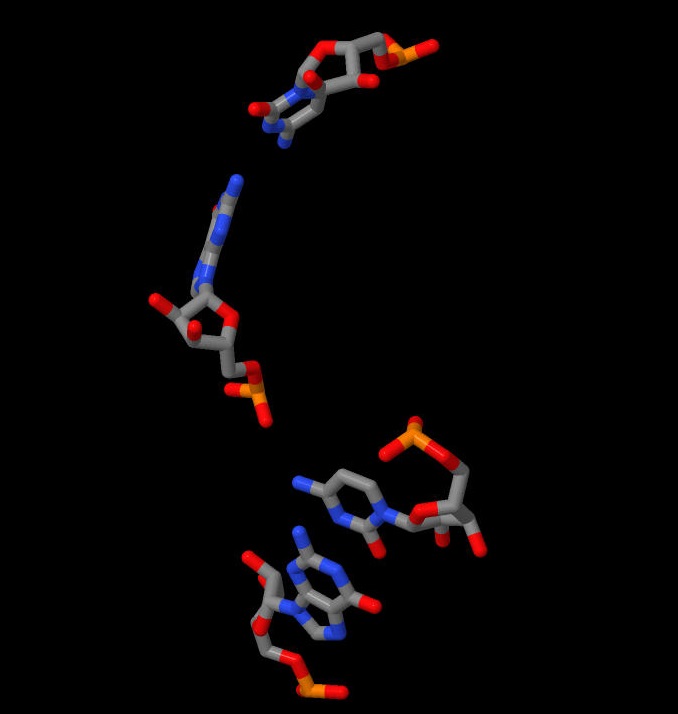 |
Источники:
| [1] Торсионные углы |