Пакет EMBOSS
В данной работе были проделаны упражнения для освоения программ из пакета EMBOSS, ниже приведён список команд и отчёт по каждому выполненному заданию.
1. Несколько файлов в формате fasta собрать в единый файл
Используемая программа - seqret. Исходные файлы: AAC74885.2.fasta, AAC74886.1.fasta, AAC74887.1.fasta, AAC74888.1.fasta, AAC74889.2.fasta, AAC74890.1.fasta, AAC74891.2.fasta, AAC74892.1.fasta, AAC74893.1.fasta. Команда:
seqret 'AAC*.fasta' join.fasta |
Ссылка на результат: join.fasta
2. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Используемая программа - seqretsplit. Исходные файлы: coding1.fasta. Команда:
seqretsplit coding1.fasta |
Ссылка на результат: aac73113.1_2.fasta, aac73114.1_3.fasta, aac73115.1_4.fasta
3. Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Используемая программа - seqret. Была выбрана последовательность полного генома Escherichia coli str. K-12 с идентификатором U00096, и из неё были выбраны три кодирующих участка, координаты которых записаны в следующем файле: mylist. Команда:
seqret @mylist l3.fasta |
Ссылка на результат: l3.fasta
4. Транслировать кодирующие последовательности в аминокислотные
Используемая программа - transeq. Исходные файлы: coding.fasta. Команда:
transeq coding.fasta 1.txt |
Ссылка на результат: pr.txt
5. Транслировать данную нуклеотидную последовательность в шести рамках
Используемая программа - transeq.
Исходные файлы: coding.fasta. Команда:
transeq coding.fasta -frame 6 6.fasta |
Ссылка на результат: 6.fasta
6. Перевести выравнивание и из fasta формате в формат .msf
Используемая программа - seqret.
Исходные файлы: alignment.fasta. Команда:
seqret alignment.fasta msf::alignment.msf |
Ссылка на результат: alignment.msf
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными
Используемая программа - infoalign.
Исходные файлы: alignment.fasta. Команда:
infoalign alignment.fasta -refseq 2 -only -name -idcount alignment.infoalign |
Ссылка на результат: alignment.infoalign
8. Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
Используемая программа - featcopy. Исходные файлы: chromosome.gb. Команда:
featcopy chromosome.gb chromosome.gff |
Ссылка на результат: chromosome.gff
9. Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product)
Используемая программа - extractfeat. Была выбрана последовательность полного генома Escherichia coli str. K-12 с идентификатором U00096, он записан в следующем файле: seq2.gb. Команда:
extractfeat seq2.gb -type CDS -describe product prot1.fasta |
Ссылка на результат: prot1.fasta
10. Перемешать буквы в данной нуклеотидной последовательности
Используемая программа - shuffleseq. Исходные файлы: coding.fasta. Команда:
shuffleseq coding.fasta shuffle.fasta |
Ссылка на результат: shuffle.fasta
11. (*) Для случайной последовательности проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (запустите blastn с порогом E = 10 - по умолчанию и посчитайте сколько с E-value < 0.1)
Используемая программа - makenucseq. Исходные файлы: нет. Команда:
makenucseq -amount 1 -length 200 random.fasta |
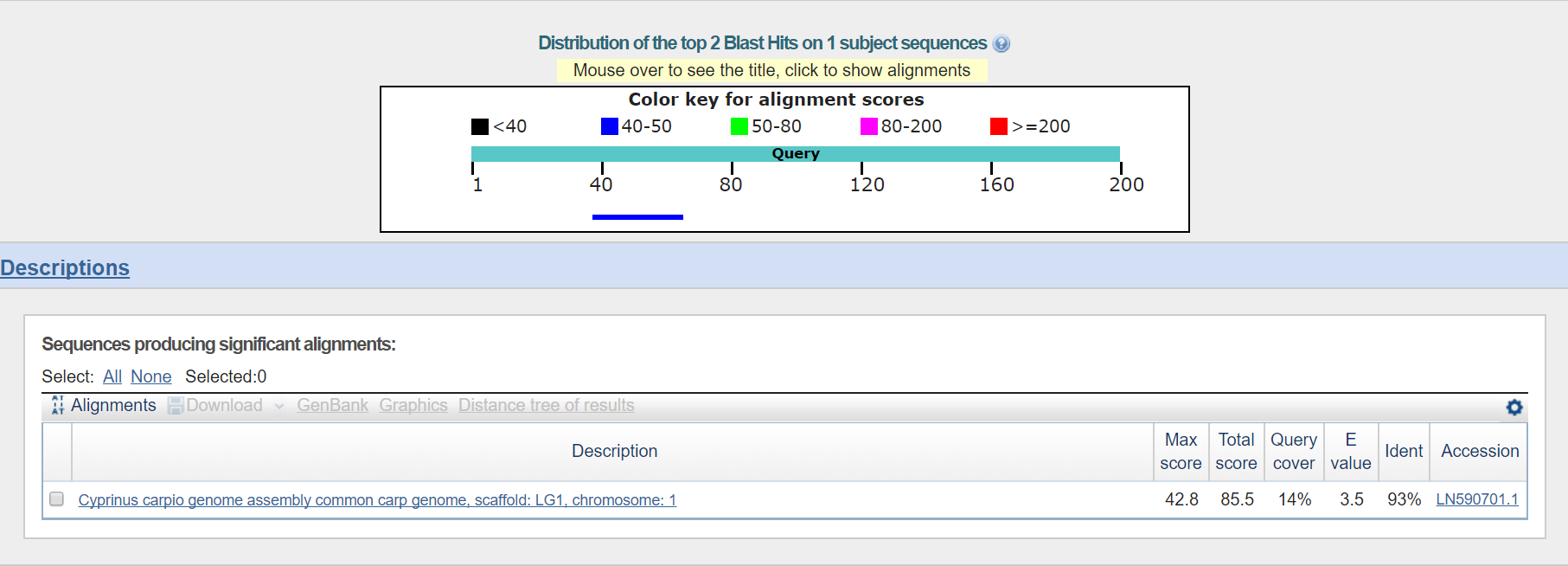
Ссылка на результат: random.fasta. Был выполнен поиск blastn с E-value 10 и найдено одно сходство с E-value = 3,5, что не является достоверным совпадением, выдача blastn представлена на Рис. 1.
 |
13. Найдите частоты кодонов в данных кодирующих последовательностях
Используемая программа - compseq. Показывает частоту встречаемости кодонов при любых рамках считывания. Исходные файлы: gene_sequences.fasta. Команда:
compseq gene_sequences.fasta -word 3 genecompseq.fasta |
Ссылка на результат: genecompseq.fasta
Используемая программа - cusp. Показывает частоту встречаемости кодонов при конкретной рамке считывания. Исходные файлы: gene_sequences.fasta. Команда:
cusp gene_sequences.fasta genecompseq.cusp |
Ссылка на результат: genecompseq.cusp
14. (*) Найдите частоты динуклеотидов в хромосоме человека, сравните их с ожидаемыми (подсказка: ожидаемая частота XY = (наблюдаемая частота X) * (наблюдаемая частота Y) )и определите динуклеотид, частота которого наиболее отклоняется от наблюдаемой
Используемая программа - compseq. Исходные файлы: chromosomey.fasta - последовательность хромосомы Y человека. Команда для получения частот встречаемости динуклеотидов:
compseq chromosomey.fasta -word 2 ydin.fasta |
Ссылка на результат: ydin.fasta Команда для получения частот встречаемости нуклеотидов:
compseq chromosomey.fasta -word 1 ydin1.fasta |
Ссылка на результат: ydin1.fasta Затем с помощью программы Excel полученные данные были обработаны и сравнены с ожидаемыми по следующей формуле: ожидаемая частота XY = (наблюдаемая частота X) * (наблюдаемая частота Y). Ссылка на файл Excel. В результате выявлено, что частота встречаемости динуклеотида TT больше всего отклоняется от ожидаемой.
15. Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
Используемая программа - tranalign. Исходные файлы: sequence.fasta - нуклеотидная последовательность, protein_alignment.fasta - белковая последовательность. Команда:
tranalign sequence.fasta protein_alignment.fasta |
Ссылка на результат: 1573620-1574456.fasta
16. Постройте локальное множественное выравнивание трех нуклеотидных последовательностей
Используемая программа - edialign. Исходные файлы: 16.3.fasta Команда:
edialign -seq 16.3.fasta -outseq align_16.fasta -outfile out_16.edialign |
Ссылка на результат: out_16.edialign и align_16.fasta
17. Удалите символы гэпов и другие посторонние символы из последовательности
Используемая программа - degapseq. Исходные файлы: gap.fasta. Команда:
degapseq gap.fasta degap.fasta |
Ссылка на результат: degap.fasta
18. Переведите символы конца строки в формат unix
Используемая программа - noreturn. Исходные файлы: coding.fasta. Команда:
noreturn coding.fasta codingunix.fasta |
Ссылка на результат: codingunix.fasta
19. Создайте три случайных нуклеотидных последовательностей длины сто
Используемая программа - makenucseq. Исходные файлы: нет. Команда:
makenucseq -amount 3 -length 100 -auto ran3.fasta |
Ссылка на результат: ran3.fasta.
20. Файл с ридами sra_data.fastq в формате fastq перевести в формат fasta
Используемая программа - seqret. Исходные файлы: sra_data.fastq. Команда:
seqret sra_data.fastq fasta::sra.fasta |
Ссылка на результат: sra.fasta.