Анализ транскриптома человека
Дано
Чтения, картирующиеся на участок хромосомы человека (получены путем секвенирования РНК), две реплики - chr2.1.fastq, chr2.2.fastq
Хромосомы человеческого генома (сборка версии hg19) - chr2.fasta
Часть I: подготовка чтений
Анализ качества чтений
С помощью программы FastQC был проведён контроль качества чтений для обеих реплик. Команды:
fastqc chr2.1.fastq fastqc chr2.2.fastq |
Получив на вход файл в формате .fastq, программа выдала архив (.zip) с отчетом по работе в виде текстового файла и рисунков, в котором помимо прочей информации находились файлы chr2.1_fastqc.html и chr2.2_fastqc.html, содержащие визуализацию качества чтений. В Таблице 1 приведены различия по чтениям первой и второй реплик.
Таблица 1. Характеристика ридов двух реплик. | ||
Категория |
chr2.1.fastq |
chr2.2.fastq |
Число ридов |
12507 |
7126 |
Длина ридов |
28-51 |
39-51 |
%GC |
46 |
44 |
На Рис. 1-2 представлена информация, полученная с помощью программы FastQC, а именно - характеристика качества каждого нуклеотида в ридах. По абсциссе расположены позиции в ридах, а по ординате - Quality score, причем данные представлены в виде диаграмм boxplot, где границами желтых столбцов служат первый и третий квартили, красная линия - медиана, синяя линия - среднее значение, а отходящие от столбцов "усы" демонстрируют размах между 10-м и 90-м процентилями. Помимо этого само поле графика покрашено в три цвета: нахождение диаграмм в зеленой плотности говорит о том, что качество данной позиции хорошее и она достоверна (выше 28), в оранжевой - среднее (от 20 до 28), в красной - плохое, таким последовательностям верить нельзя (от 0 до 20).
|
|
Рис. 1. Характеристика качества последовательностей ридов первой реплики. |
Рис. 2. Характеристика качества последовательностей ридов второй реплики. |
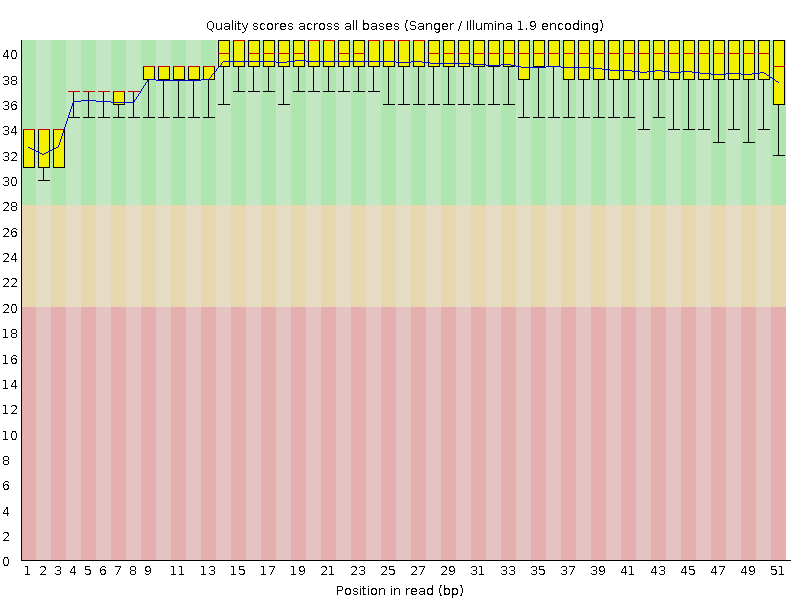

Можно заметить, что качество последовательностей ридов примерно одинаковое в обеих репликах, все диаграммы входят входят в зелёную область, следовательно, для всех позиций в чтениях quality score > 28, что я является очень хорошим показателем. Помимо этого, в html-файлах содержится ещё много полезной информации, ниже приведен анализ некоторых графиков, выдаваемых программой. На Рис. 3-4 представлена тепловая карта качества последовательностей, поступающих из проточных ячеек в Illumina - такую информацию можно получить только для этого метода секвенирования. Здесь по вертикальной оси обозначены номера ячеек, а по горизонтальной - позиции в риде. Синий цвет характеризует хорошее качество, а градация к красному демонстрирует падение качества позиции.
|
|
Рис. 3. Качество проточных ячеек для первой реплики. |
Рис. 4. Качество проточных ячеек для второй реплики. |
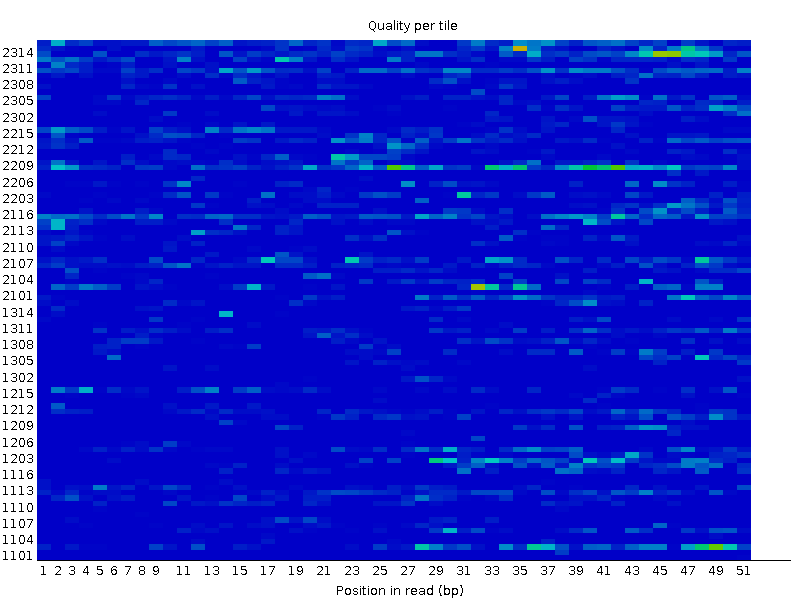
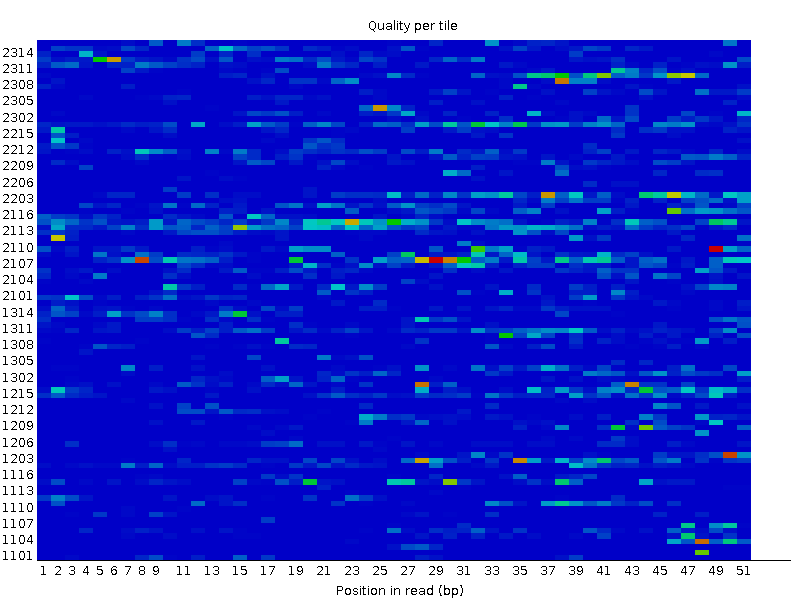
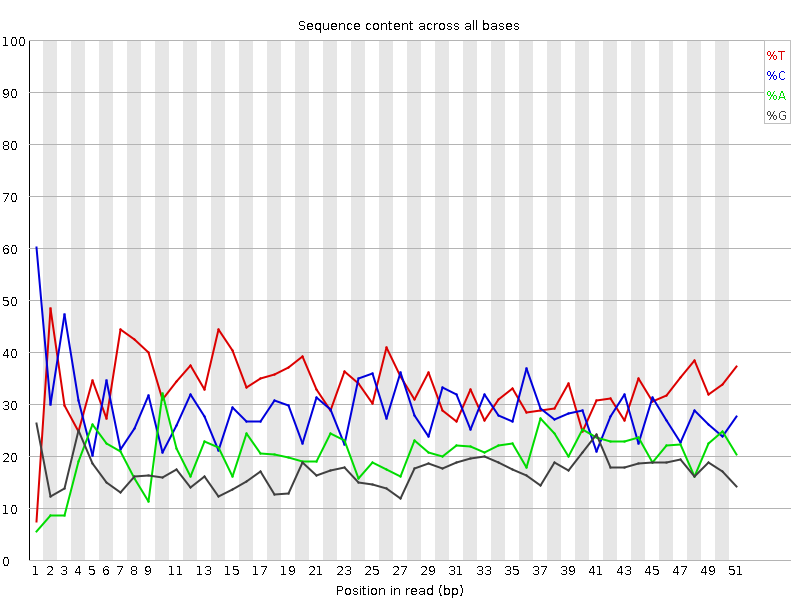
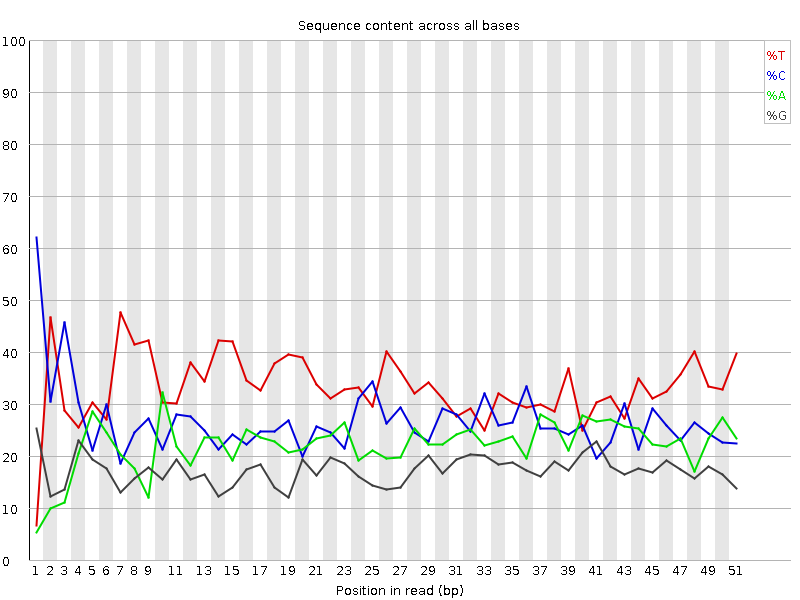
Как можно заметить, у второй реплики большее количество ячеек с позициями с низким качеством и встречаются даже красные, означающие очень низкое качество позиции в чтении, ячейки. В следствие чего программа указала, что нужно обратить внимание на эти данные для первой реплики, а для второй, что эти данные могут вносить какие-то помехи. Также программа FastQC выдает частоту встречаемости азотистых оснований для каждой позиции в ридах. В хорошем секвенировании частота встречаемости оснований должны быть постоянными из позиции в позицию. На Рис. 5-6 представлена такая характеристика, можно заметить, что в обеих репликах на протяжении всех позиций наблюдаются скачки частот, возможно, это может быть объяснено наличием оверэкспрессированных участков, в частности 200 таких участков найдены в первой реплике и 179 - во второй.
|
|
Рис. 5. Частота встречаемости азотистых оснований для первой реплики. |
Рис. 6. Частота встречаемости азотистых оснований для второй реплики. |
Часть II: картирование чтений
Картирование чтений
С помощью программы Hisat2 было осуществлено картирование чтений. Сначала была проиндексирована референсная последовательность с помощью команды:
hisat2-build chr2.fasta chr2 |
В результате чего были получены 8 файлов в формате .ht2. Затем построено выравнивание обеих реплик и референса в формате .sam с помощью команды, где параметр --no-softclip запрещает подрезать чтения:
hisat2 -x chr2 -U chr2.1.fastq --no-softclip > align.sam hisat2 -x chr2 -U chr2.2.fastq --no-softclip > align2.sam |
Примечание: В отличие от предыдущего практикума при выполненнии этой команды был убран параметр --no-spliced-alignmen, так как анализируется последовательности РНК, подвергающиеся сплайсингу, поэтому в данном случае разрывы можно разрешить. В результате выдача сохранена в файл align.sam - для первой реплике, align2.sam - для второй реплики.
Анализ выравнивания
С помощью команды, приведенной ниже, выравнивания из формата .sam было переведено в бинарный формат .bam:
samtools view align.sam -bo align1.bam samtools view align2.sam -bo align2.bam |
Для этого использовалась команда view из пакета samtools. Затем выравнивания чтений с референсом было отсортировано по координате в референсе начала чтения с помощью следующей команды:
samtools sort align1.bam -T 1.txt -o chr2.1.bam samtools sort align2.bam -T 1.txt -o chr2.2.bam |
И далее отсортированные файлы chr2.1.bam и chr2.2.bam были проиндексированы командой:
samtools index chr2.1.bam samtools index chr2.2.bam |
После чего получены файлы chr2.1.bam.bai и chr2.2.bam.bai. Затем выяснялось, сколько чтений откартировано на геном, эта информация выдавалась сразу после работы программы Hisat2, в Таблицу 2 сведены эти данные.
Таблица 2. Число откартированных ридов для обеих реплик. | ||
chr2.1 |
chr2.2 | |
12507 reads; of these:
12507 (100.00%) were unpaired; of these:
82 (0.66%) aligned 0 times
12377 (98.96%) aligned exactly 1 time
48 (0.38%) aligned >1 times
99.34% overall alignment rate
|
7126 reads; of these:
7126 (100.00%) were unpaired; of these:
46 (0.65%) aligned 0 times
7056 (99.02%) aligned exactly 1 time
24 (0.34%) aligned >1 times
99.35% overall alignment rate
|
|
| Из 12507 чтений 82 не откартировались ни разу, 48 были откартированы больше, чем один раз, а все остальные (12377) - ровно один раз. | Из 7126 чтений 46 не откартировались ни разу, 24 - больше, чем один раз, а 7056 - ровно 1 раз. | |
samtools idxstats chr2.1.bam > map1.txt samtools idxstats chr2.2.bam > map2.txt |
В результате в файлах map1.txt и map2.txt была записана следующая информация: длина референсной последовательности - 243199373 и число откартированных чтений, однако никаких пояснений не даётся.
С помощью программы IGV - Integrative Genomics Viewer, были визуализированы выравнивания обеих реплик. На вход программа получает отсортированный файл с выравниванием в формате .bam. Рис. 7 демонстрируют результат работы этой программы, причем она сразу способна отобразить оба выравнивания относительно одного референса. Верхней линией на рисунках показана картированная хромосома, что позволяет довольно легко найти нужный участок.
|
Рис. 7. Визуальзация выравнивания реплик. |
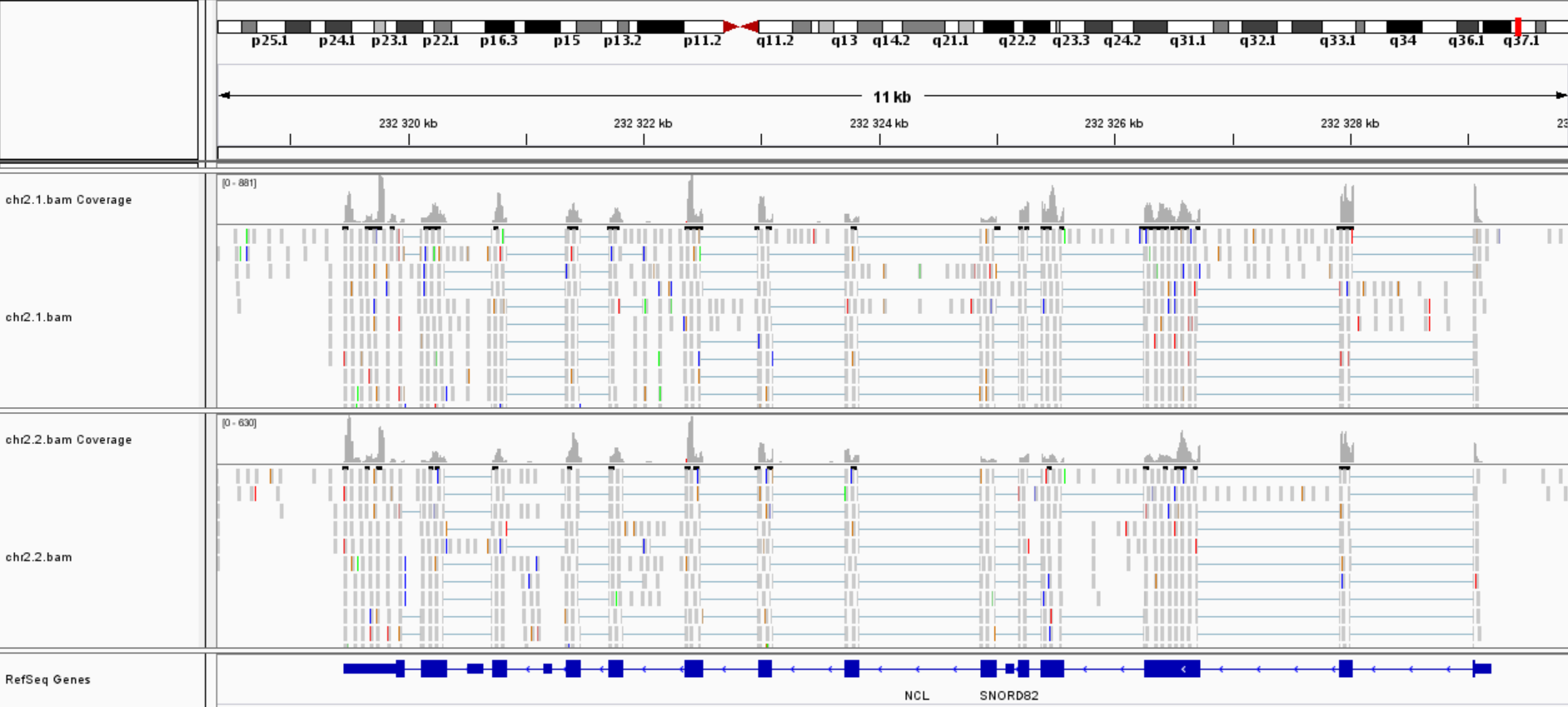
Можно заметить, что отличие такого выравнивания от геномных чтений, рассматриваемых в предыдущем практикуме в том, что длина ридов намного меньше длины ридов для геномов. Также видно, что гены намного более полно покрываются ридами, причем чтения в основном покрывают кодирующие участки генов. Полосами показывается то, что рид приходится на кодирующие участки, получающиеся на мРНК в результате вырезания участка, изображенного полосой.
Часть III: Работа с Bedtools
Подсчет чтений
С помощью программы Bedtools был выполнен перевод полученных выравниваний из формата .bam в формат .bed, с которым способна легко работать данная программа, это выполнено для обеих реплик. Команды:
bedtools bamtobed -i chr2.1.bam >2.1.bed bedtools bamtobed -i chr2.2.bam >2.2.bed |
Далее с помощью программы intersect производилось пересечение реплик с известной разметкой генов с помощью команд, где параметр -a отражает разметку, с которой выполняется пересечение, а -b - реплику, -с проводит подсчет количества ридов, перекрывающих ген:
bedtools intersect -a gencode.genes.bed -b 2.1.bed -c > coun2.1.bed bedtools intersect -a gencode.genes.bed -b 2.2.bed -c > coun2.2.bed |
В результате были получены два файла coun2.1.bed и coun2.2.bed, которые затем были отсортированы по 6 колонке, где и содержалась информация о присутствии чтений, покрывающих гены. Команды:
sort -k 6 -r coun2.1.bed > sort2.1.bed sort -k 6 -r coun2.2.bed > sort2.2.bed |
Информация о генах, в которые вошли чтения из обеих реплик, сведена в Таблицу 3.
Таблица 3. Характеристика генов, в которые попали риды обеих реплик. | |||||
chr2.1 |
chr2.2 | ||||
Ген |
Количество чтений |
Информация о гене |
Количество чтений |
||
NCL |
111 |
Ген, кодирующий белок нуклеолин, расположенный в локусе 2q37.1, - один из наиболее часто встречающихся ядрышковых белков. Он принимает участие в процессе образования рибосом, но выполняет также функции, не имеющие прямого отношения к ядрышку и протекающему в нём биогенезу рибосом. Способность нуклеолина участвовать во многих клеточных процессах обеспечивается его структурной организацией и возможностью взаимодействовать со многими белками, а также нуклеиновыми кислотами. Нуклеолин играет определённую роль в развитии различных вирусных инфекций, а также при возникновении раковых заболеваний, выступая как онкоген [1]. |
111 |
||
SNORD20 | 3 |
Последовательность малой ядерной РНК, представляющей собой некодирующую РНК, функцией которой является модификация других малях ядерных РНК. Кодируется в 11 интроне гена нуклеолина у человека, мыши и крысы [2]. | 3 |
||
AC017104.2 | 4 |
Псевдоген с координатами: 231437101-231437421 [3]. | 2 |
||
SNORA75 | 3 |
Малая ядерная РНК, связывающая ряд белков, формирующих малый ядерный РНК-белковый комплекс. Считается, что данная РНК модифицирует уридиы в псевдоуридины, непосредственно взаимодействуя с РНК, рибосомальными, спайсосомальные и другие. Кодируется внутри интрона 12 гена нуклеолина у человека, мыши и других организмов [4]. |
3 |
||
SNORD82 | 3 |
Малая ядерная РНК, участвующая в модификации прерибосомальной РНК, а именно в её метилировании [5]. | 3 |
||
Ген |
Информация о гене |
||||
TBC1D8 |
Кодирует белок, обладает 10 транскриптами (за счет разного пути сплайсинга), 82 ортолога и 7 паралогов, может выступать в качестве ГТФаз-активирующего белка для белков из семейства Rab [6]. | 5 |
|||
Изучение опций Bedtools
1. Получите из файла в выравниванием файл с чтениями в формате fastqБыл взят файл, полученный выше в формате .bam и переведен в fastq-формат. Команда:
bedtools bamtofastq -i chr2.1.bam -fq chr2.1.fq |
2. Получите файл с нуклеотидной последовательностью (.fasta) для одного из покрытых Вашими чтениями генов
Для выполнения данного задания был создан файл 1.bed с координатами одного из генов, который покрывается ридами, информация об этом приведена выше. А затем из полной последовательности хромосомы 2 человека была получена его последовательность и выдача записана в файл gene.fasta в fasta-формате. Команда:
bedtools getfasta -fi chr2.fasta -bed 1.bed > gene.fasta |
3. Разбейте свою хромосому на фрагменты по 1 млн нуклеотидов
В данном задании на вход программа получала файл chr2.txt, в котором была записана длина хромосомы, равняющаяся 242193529 п.н., а затем программа разбивала хромосому на интервалы длиной 1000000 п.н. и записывала свою выдачу в файл wind.txt. В результате работы программы получилось 243 интервала. Команда:
bedtools makewindows -g chr2.txt -w 1000000 > wind.txt |
4. Объедините Ваши чтения в кластеры
При выполнении данной команды на вход поступает файл с сортированными выравненными чтениями в формате .bed, параметр -d позволяет объединять чтения, находящиеся на расстоянии 5 п.н. и ближе, в результате получен файл с объединением в кластеры cluster.txt. Команда:
bedtools cluster -i 2.1.bed -d 5 > cluster.txt |
5. Наберите из Вашей хромосомы 1000 случайных фрагментов по 200 нуклеотидов
В данном задании на вход программа получала файл chr2.txt, в котором была записана длина хромосомы, равняющаяся 242193529 п.н., а затем программа сгенерировала 1000 рандомных последовательностей длиной 200 п.н., а выдача программы записывалась random.txt. Команда:
bedtools random -g chr2.txt -n 1000 -l 200 > random.txt |
7. Получите координаты одного из покрытых Вашими чтениями генов, расширенные на 1000 нуклеотидов в обе стороны
Для выполнения данного задания был создан файл 1.bed с координатами одного из генов, который покрывается ридами, информация об этом приведена выше. С помощью программы slop был получен файл slop.txt, в котором записаны координаты гена на 1000 п.н. больше слева и справа от него. Команда:
bedtools slop -i 1.bed -g chr2.txt -b 1000 > slop.txt |
8. Получите координаты одного из покрытых Вашими чтениями генов, сдвинутые на 500 нуклеотидов ближе к началу хромосомы
На вход программа получала всё те же файлы с координатами гена и с указанным числом пар нуклеотидов в хромосоме, а её выдача записывалась в файл shift.txt, в котором указывались координаты гена, сдвинутые на 500 п.н. ближе к началу хромосомы. Команда:
bedtools shift -i 1.bed -g chr2.txt -s -500 > shift.txt |
9. Получите непересекающиеся фрагменты, соответствующие области, покрытой Вашими чтениями
Если подразумевается получение фрагментов, которые не пересекаются с другими чтениями, то после получения файла cov.txt (см. Пункт 10), была выполнена команда, достающая из этого файла строчки, где пересечение равно 1, и получаем файл с координатами таких участков cov1.txt. Команда:
grep -w 1 cov.txt > cov1.txt |
Если же подразумевается получение участков, непересеченных ридами, то с помощью программы complement можно получить координаты участков хромосомы, непересечнной ридами, выдача программы записывается в файл compl.txt:
bedtools complement -i 2.1.bed -g chr2.txt > compl.txt |
10. Получите файл с координатами интервалов, покрытых Вашими чтениями, с информацией о покрытии в любом формате
На вход программа получала файл с сортированным выравниванием чтений и длиной хромосомы, а на выходе получется файл с координатами интервалов, покрытых чтениями - cov.txt, где в 4 столбце представлено количество чтений, покрывающий данный промежуток. Команда:
bedtools genomecov -i 2.1.bed -g chr2.txt -bg > cov.txt |
Таблица 7. Команды использованные для выполнения данного практикума. | ||||||||
Команда |
Пояснение |
|||||||
fastqc chr2.1.fastq fastqc chr2.2.fastq |
Проводит контроль качества чтений |
|||||||
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Доступ к файлам для запуска программы Hisat2 |
|||||||
hisat2-build chr2.fasta chr2 |
Индексирование референсной последовательности, выдает файлы в формате .ht2 |
|||||||
hisat2 -x chr2 -U chr2.1.fastq --no-softclip > align.sam hisat2 -x chr2 -U chr2.2.fastq --no-softclip > align2.sam |
строит выравнивание очищенных чтений и референса в формате .sam. Параметр --no-softclip запрещает подрезать чтения. |
|||||||
samtools view align.sam -bo align1.bam samtools view align2.sam -bo align2.bam |
Переводит выравнивание в бинарный формат |
|||||||
samtools sort align1.bam -T 1.txt -o chr2.1.bam samtools sort align2.bam -T 1.txt -o chr2.2.bam |
Сортирует выравнивание чтений с референсом в формате .bam по координате в референсе начала чтения, в файл 1.txt записана выдача stdout после работы программы |
|||||||
samtools index chr2.1.bam samtools index chr2.2.bam |
Индексирует сортированный файл |
|||||||
samtools idxstats chr2.1.bam > map1.txt samtools idxstats chr2.2.bam > map2.txt |
Выводит информацию об откартированных чтениях на геном |
export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin |
Доступ к файлам для запуска программы Bedtools |
|||||
bedtools bamtobed -i chr2.1.bam >2.1.bed bedtools bamtobed -i chr2.2.bam >2.2.bed |
Переводит файл из формата .bam в формат .bed |
|||||||
bedtools intersect -a gencode.genes.bed -b 2.1.bed -c > coun2.1.bed bedtools intersect -a gencode.genes.bed -b 2.2.bed -c > coun2.2.bed |
Пересекает разметку генов с репликами |
|||||||
sort -k 6 -r coun2.1.bed > sort2.1.bed sort -k 6 -r coun2.2.bed > sort2.2.bed |
Сортирует файл по 6 толбцу по убыванию |
|||||||
Источники:
| [1]Нуклеолин [2]SNORD20 [3]AC017104.2 [4]SNORA75 [5]SNORD82 [6]TBC1D8 |