

Выбор семейства домена
В качестве семейства доменов для дальнейшей работы был выбран домен Poly A polymerase head domain (домен поли(А)-полимеразной активности). Его описание приведено в таблице 1.
| AC | ID | Функция домена | Число архитектур | Число последовательностей | Число видов |
|---|---|---|---|---|---|
| PF01743 | PolyA_pol | Добавляет поли(А)-хвост к 3'-концу многих РНК, который обычно является меткой для деградации. | 54 | 6516 | 4623 |
Для выбранного домена с Pfam было скачано выравнивание всех доступных последовательнотей с этим доменом - представлено в проекте Jalview и fasta-файле. В качестве окраски использовалась стандартная ClustalX с порогом консервативности 10%. К последовательности C9QS13_ECOD1 бактерии Escherichia coli была прикреплена её 3D-структура 3AQK.
Выбор архитектур
Выбранный домен входит в 54 различные архитектуры, просмотреть которые пожно по ссылке. В качестве рассматриваемых архитектур были выбраны архитектуры, указанные в таблице 2.
| Архитектура | Изображение | Число представителей | Характеристика смежных доменов |
|---|---|---|---|
| PolyA_pol, PolyA_pol_RNAbd, HD | 
| 1943 | PolyA_pol_RNAbd (PF12627) - возможный сайт связывания РНК с поли(А)-полимеразой HD (PF01966) - домен металл-зависимой фосфогидролазной активности |
| PolyA_pol, PolyA_pol_RNAbd, tRNA_NucTran2_2 | 
| 1450 | tRNA_NucTran2_2 (PF01966) - домен нуклеотидилтрансферазной активности (обеспечивает присоединение ССА к 3'-концу) |
Выбор таксона и подтаксонов
В качестве таксона для поиска представителей архитектур былы выбраны бактерии (Bacteria). Соотвественно в качестве подтаксона следующего уровня выбраны протеобактерии (Proteobacteria) и фирмикуты (Firmicutes). Такой выбор был сделан исходя из распределения домена по таксонам, приведённом также на рис. 1.
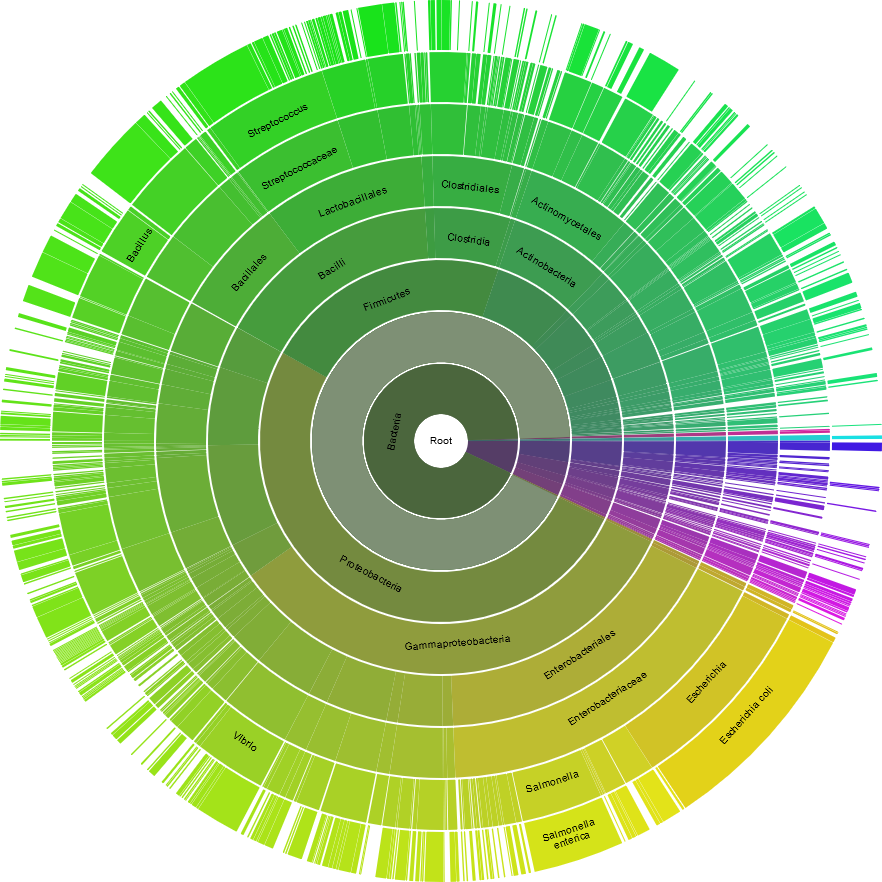
Рис. 1 Распределение домена Poly A polymerase head domain по таксонам.
Выбор представителей архитектур
С помощью скрипта swisspfam_to_xls.py была получена таблица (Excel, лист "scrypt_1") с информацией об архитектуре всех последовательностей, содержащих домен Poly A polymerase head domain. Для этого он был запущен на сервере Kodomo коммандой: python swisspfam-to-xls.py -z -p PF01743
После этого получен список последовательностей с указанием доменной архитектуры. Для этого построена сводная таблица (Excel, лист "Сводная seq_id-pfam_ac"), где по строкам расположены ID последовательностей, а по столбцам – домены Pfam.
Затем с сервера Uniprot были загружены полные записи всех последовательностей, AC которых имеются в сводной таблице. С помощью скрипта uniprot_to_taxonomy.py из этих записей была извлечена таксономия организмов (Excel, лист "taxonomy"). После этого таксономия перенесена в основную таблицу (Excel, лист "Общая").
На листе "Сводная" Excel расположена сводная таблица, в которой напротив AC последовательностей находятся домены рассматриваемых архитектур. С помощью включённого фильтра можно отбирать последовательности определённых таксонов (соответственно протеобактерий или фирмикут). Таким образом было выбрано по 20 последовательностей из каждой архитектуры, по 10 для каждого филума. Все отобранные ID были проверены на сервере Pfam на принадлежность к нужной архитектуре, т.к. данный отбор не исключает возможность присутствия дополнительных доменов. Но т.к. рассматриваемые архитектуры являются наиболее распространёнными, то метод оправдывает себя для этой задачи. Отобранные ID представлены в файле.
Для удобства были введены сокращения, указанные в таблице 3.
| Архитектура 1
| Архитектура 2
| |
| Proteobacteria | Pro1 | Pro2 |
| Firmicutes | Fir1 | Fir2 |
После с помощью скрипта filter-alignment.py из уже приводившегося выравнивания были вырезаны последовательности, соответствующие выбранным ID. Это выравнивание представлено на рис. 2.
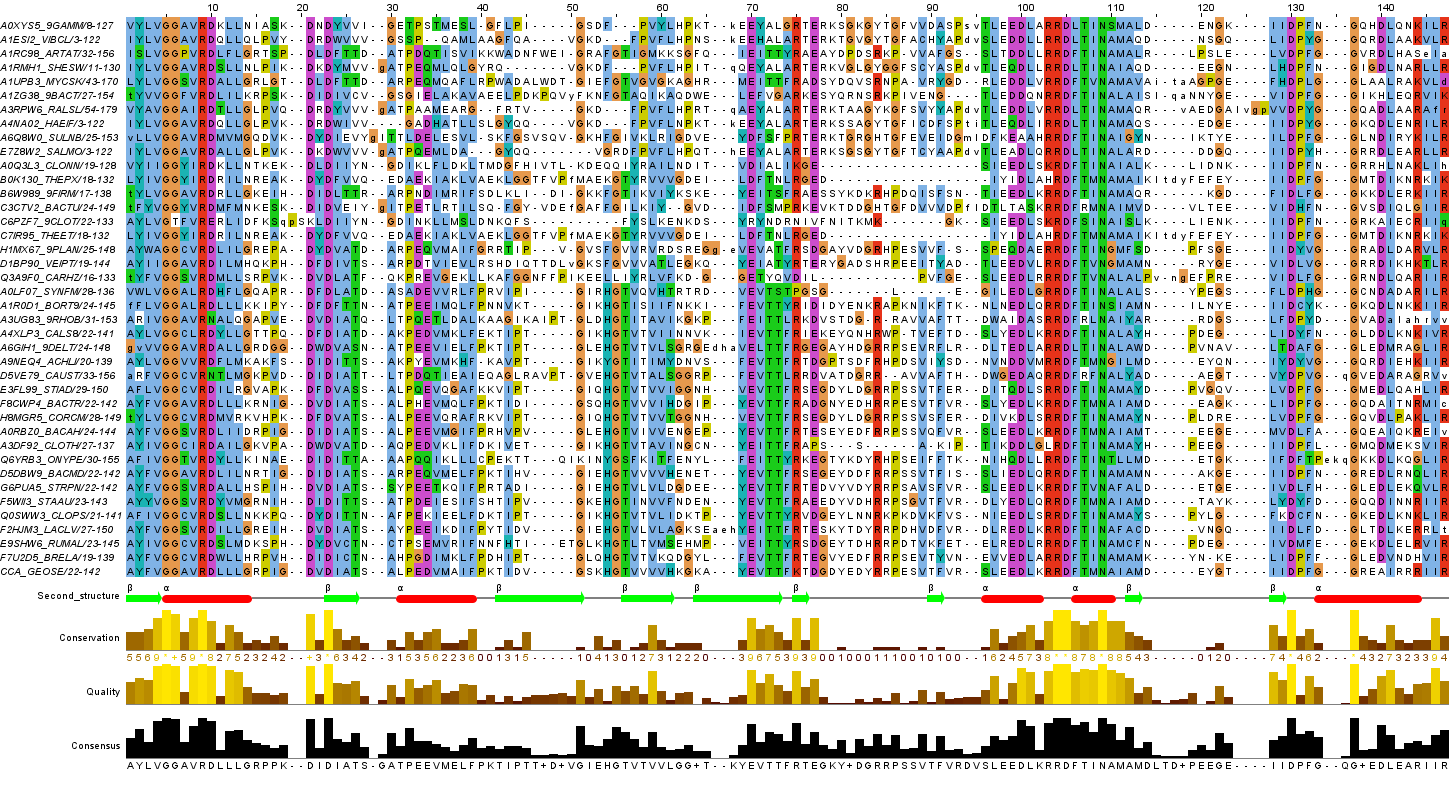
К этому выравниванию была добавлена пространственная структура белка CCA_GEOSE (1MIV.pdb), по которой была добавлена строка аннотации "Second_structure". Также выравнивание было отредактировано: удалены пустые столбцы, перенесены аминокислоты для заполнения гэпов во вторичных структурах, проведена сортировка: наверх подняты последовательности с архитектурой 1, вниз опущены - с архитектурой 2. Конечый вариант представлен в проекте Jalview и на рис. 3.

Рис. 3 Отредактированное выравнивание последовательностей белков из выборки. Окраска: ClustalX с порогом консервативности 15%. Серым выделена последовательность с 3D-структурой.
Построение филогенетического дерева
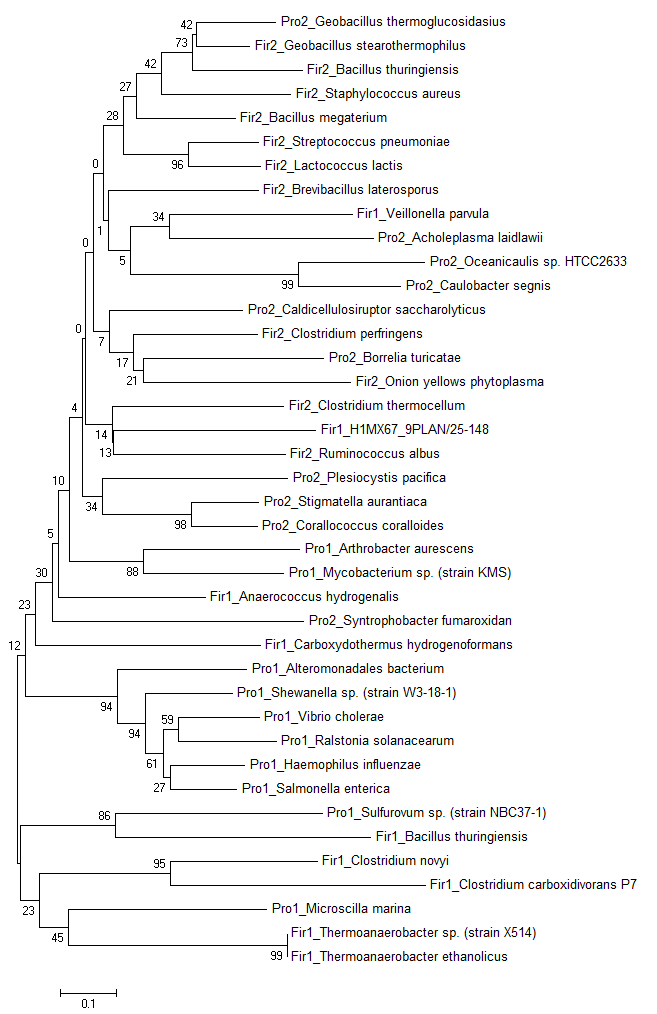
С помощью программы MEGA методом Neighbor-Joining было построено филогенетическое дерево по выравниванию выборки белков с доменом Poly A polymerase head domain - рис. 4 (с bootstrap).
{kind=link}

Рис. 4 Филогенетическое дерево белков из выборки.
Скобочное представление дерева доступно в newick-файле. С помощью сервиса ITOL полученное дерево было раскрашено и переведено в круговую форму (рис. 5).
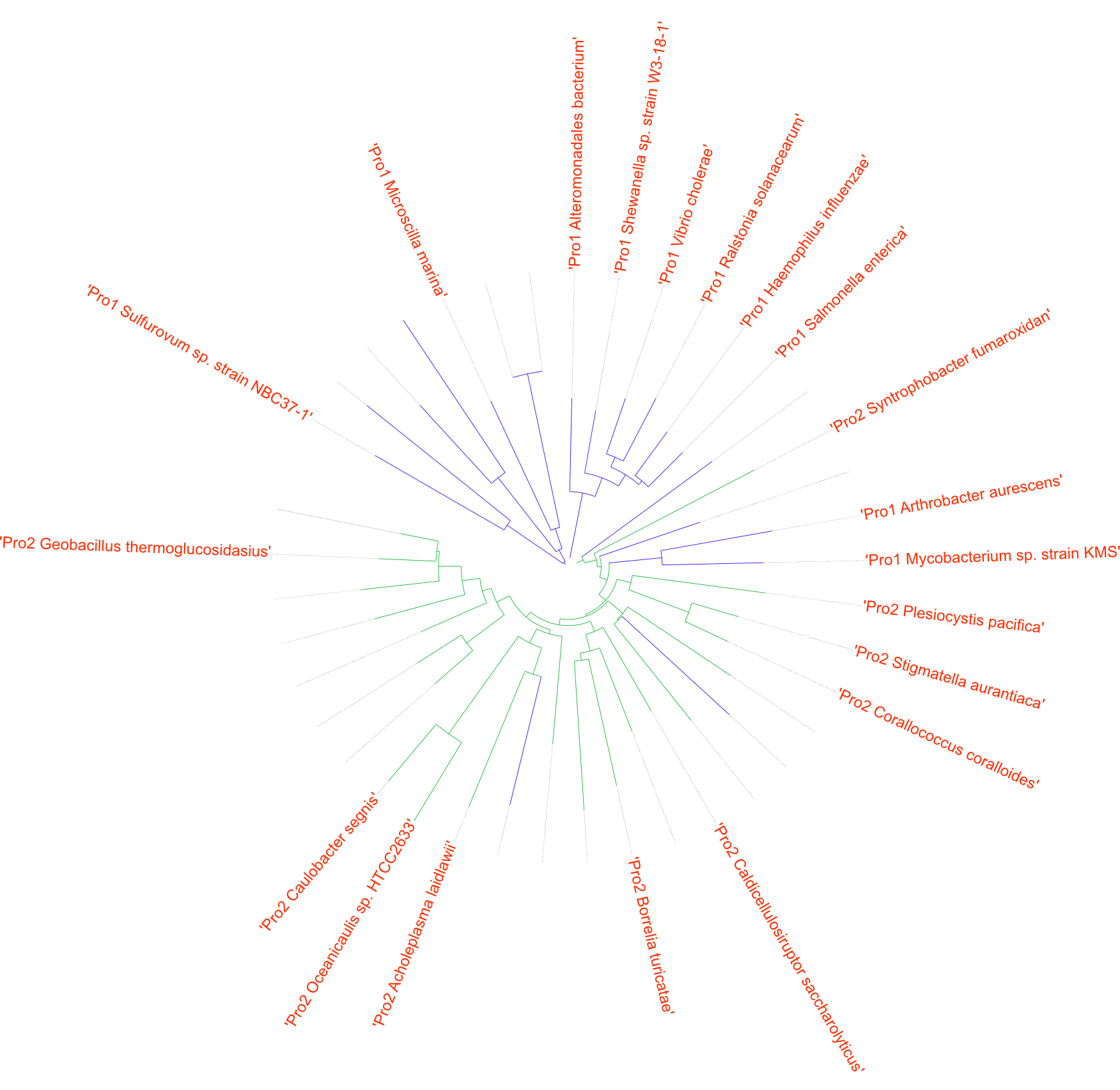
Рис. 5 Филогенетическое дерево белков из выборки. Обозначения: синий - архитектура 1, зелёный - архитектура 2, красный - протеобактерии, белый - фирмикуты.
Обсуждение
Согласно рисунку 5, рассматриваемые архитектуры разошлись очень рано (деление на зелёный и синий происходит в самом корне), после чего происходило развитие таксонов. Даже на выравнивании на рис. 3 видно, что домены двух различных архитектур имеют разноую последовательность, т.к. в архитектуре 1 центральный фрагмент почти не консервативен, по сравнению с архитектурой 2.
Примечательно, что не все ветви имеют хорошую поддержку. Так, ветви, которые делят дерево на крупные клады имеют очень маленькую bootstrap-поддержку. Однако мелкие клады имеют высокую поддержку.