Построение совмещения структур
На сервере EBI с помощью PDBeFold был проведён поиск структурных гомологов цепи А белка 1XTM (мутант SOD-like белка из B. subtilis) с параметрами по-умолчанию. Из результатов выдачи было выбрано 5 структурных гомологов (см. табл. 1).
| Белок | ID | Nalign | RMSD | Q-score | |
|---|---|---|---|---|---|
| 1 | Нативная SOD B.subtilis | 1s4i:C | 152 | 0.43 | 0.98 |
| 2 | Cu/Zn SOD гидры | 4oja:A | 134 | 1.62 | 0.61 |
| 3 | SOD-1 быка | 1e9o:A | 133 | 1.53 | 0.61 |
| 4 | SOD хлоропласта томата | 3km2:J | 132 | 1.45 | 0.61 |
| 5 | Cu/Zn SOD Xenopus laevis | 1xso:A | 133 | 1.57 | 0.61 |
С помощью того же сервиса PDBeFold было проведено множественное выравнивание выбранных гомологов и исходного белка. Результаты представлены в таблице 2, также доступны совмещение структур (см. рис. 1) и выравнивание последовательностей  (см. рис. 2). По значениям RMSD и Q-score сразу бросается в глаза, что SOD-like белок бактерии отличается от остальных SOD белков.
(см. рис. 2). По значениям RMSD и Q-score сразу бросается в глаза, что SOD-like белок бактерии отличается от остальных SOD белков.
| Белок | ID | Nalign | RMSD | Q-score | |
|---|---|---|---|---|---|
| 0 | Мутант SOD B.subtilis | 1xtm:A | 152 | 1.0319 | 0.7765 |
| 1 | Нативная SOD B.subtilis | 1s4i:C | 151 | 1.1815 | 0.7568 |
| 2 | Cu/Zn SOD гидры | 4oja:A | 149 | 0.7752 | 0.8305 |
| 3 | SOD-1 быка | 1e9o:A | 151 | 0.6321 | 0.8370 |
| 4 | SOD хлоропласта томата | 3km2:J | 153 | 0.6587 | 0.8231 |
| 5 | Cu/Zn SOD Xenopus laevis | 1xso:A | 150 | 0.6677 | 0.8385 |

Рисунок 1. Выравнивание гомологов белка 1XTM. Совпадает практически всё, кроме двух петель, указанных стрелками.

Рисунок 2. Выравнивание последовательностей гомологов по структурному выравниванию.
Также было проведено множественное выравнивание последовательностей выбранных белков программой MUSCLE на сервере EBI (см. рис. 3), результаты которого представлены в виде файла .

Рисунок 3. Выравнивание последовательностей гомологов с помощью MUSCLE.
Данные выравнивание почти совпадают, но различаются по длине и положением гэпов (см. рис 4).
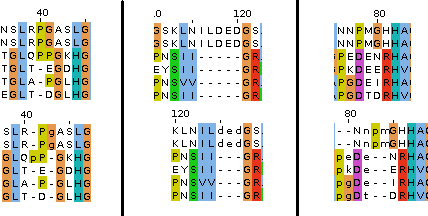
Рисунок 4. Примеры различного положения гэпов в выравнивании по последовательности (сверху) и по структуре (снизу): неправильное выравнивание в 3 нижних последовательностях (слева), различная длина гэпа (по центру) наличие/отстутствие гэпа (справа).
Первое несоответствие было разобрано подробнее. На рисунке 5 выделен столбец 42 в нумерации выравнивания по структурам (PPPEPD). Во-первых, все выделенные аминокислоты почти совпадают, поэтому они должны находиться в одном столбце. Во-вторых, правее от них выступают 2 глицина (указаны стрелочкой), а потом полипептидная цепь встаёт "на место". Поэтому эти глицины должны находиться в отдельном столбце. В-третьих, левее от этого столбца выпирают пролин и глутамин (также указаны стрелочкой), которые соответствуют одной аминокислоте в остальных последовательностях. Отразить такую деталь на выравнивании букв, дело не тривиальное. Как результат, в данном случае, по всем трём пунктам выигрывает выравнивание по структурам, что не удивительно.
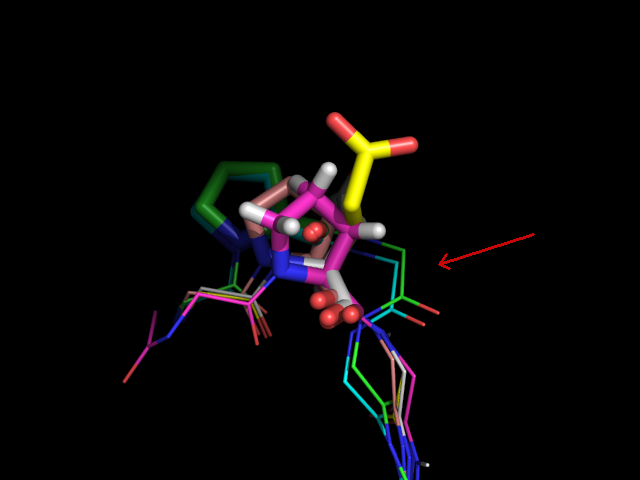
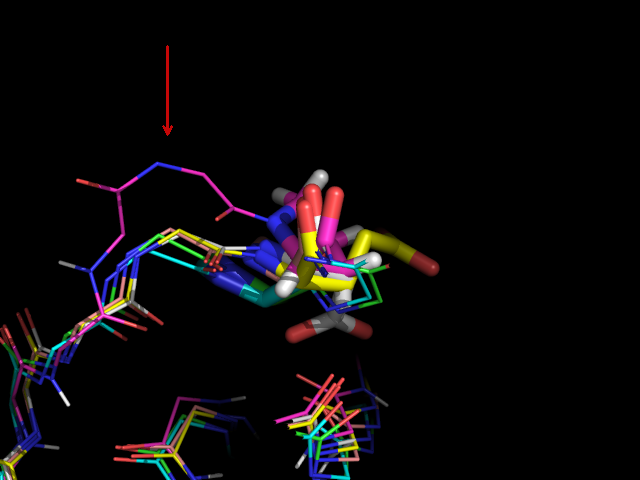
Рисунок 5. Демонстрация выравнивания из рисунка 4 слева. Подробное описание в тексте.
Поиск по сходству
С помощью PDBeFold был проведён поиск с параметрами по умолчанию домена 1B0M A:203-315 (см. рис. 6). Ожидается, что среди первых результатов (37) по RMSD должна быть сама структура 1B0M, однако её там нет. Причина заключается в том, что по умолчанию установлен параметр, отвечающий за то, чтобы сравниваемая структура была похожа на исходную не менее, чем на 70%. А как видно из рисунка 6, домен, по которому ведётся поиск составляет 15-20% от всей структуры, от чего и нет структуры 1B0M среди результатов выдачи. При запуске с порогом 15% эта структура находится, с RMSD 0.00 (среди 370 хитов).
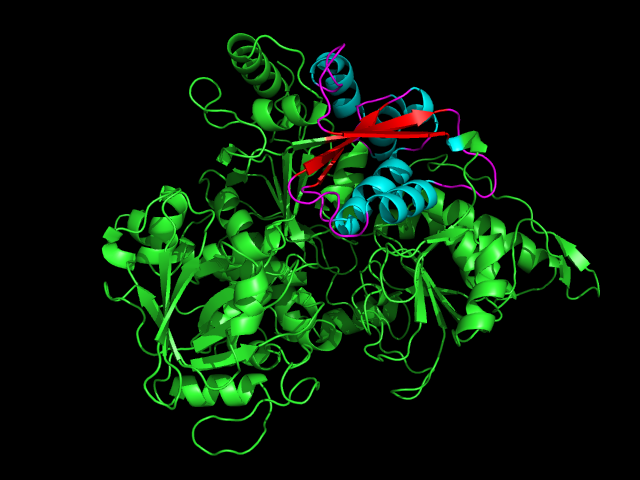
Рисунок 6. Домен A:203-315 в структуре 1B0M.
Совмещение по заданному выравниванию
Было выбрано 2 структуры константного домена T-клеточного рецептора из цепочки альфа (2BNQ D:115-204) и из цепочки бета (1BD2 E:119-247).
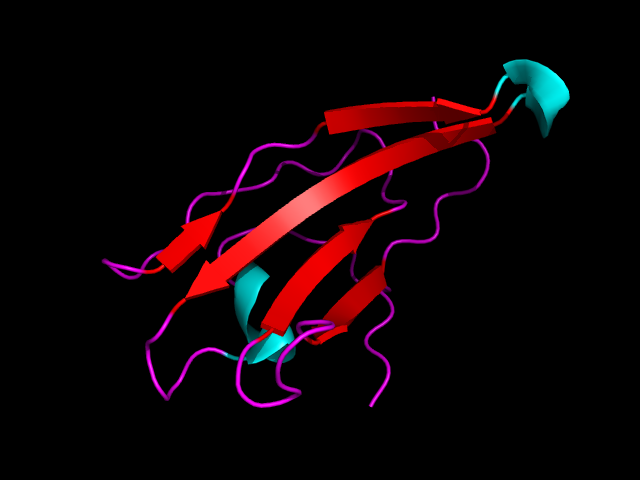
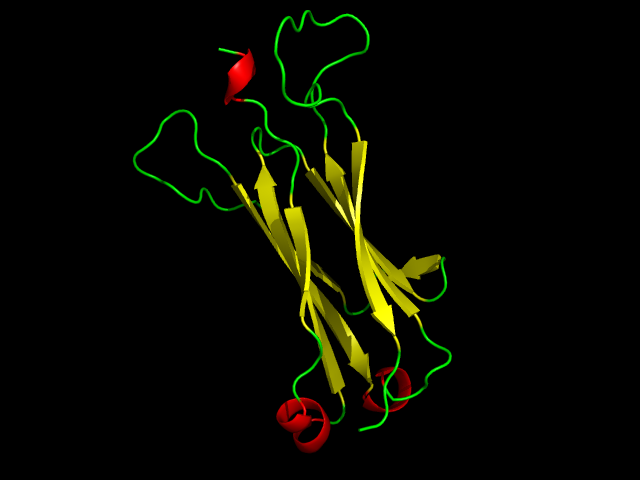
Рисунок 7. Константные домены Т-клеточного рецептора человека из цепочки альфа (слева) и бета (справа).
Для каждого домена были построены карты β-листов с помощью SheeP (см. рис. 8-9). Карта sht_0 структуры 2BNQ скорее всего соответсвует карте sht_0 структуры 1BD2, расположенной в той же ориентации.
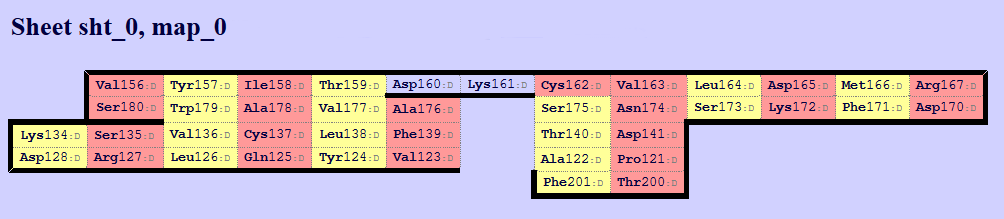
Рисунок 8. Карта β-листа домена структуры 2BNQ.
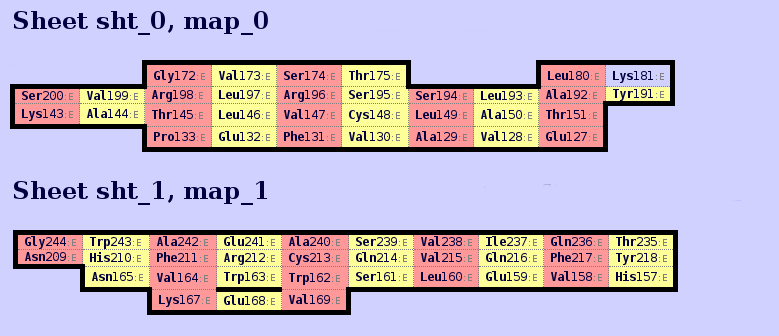
Рисунок 9. Карта β-листов домена структуры 1BD2.
В каждом листе присутствует консервативный остаток цистеина, Cys137 (образует дисульфидный мостик с Cys187) в 2BNQ и Cys148 (образует мостик с Cys213) в 1BD2, соответствующие друг другу, т.к. оба окружены остатками валина и лейцина. С помощью команды pair_fit эти структуры были совмещены в PyMol по 4 тяжам и 3 гребням, окружающие указанные цистеины (см. рис. 10):
select sel1, chain D and resi 157-158+177-179+136-138+124-126 and name CA
select sel2, chain E and resi 174-175+194-196+147-149+129-131 and name CA
pair_fit sel1, sel2
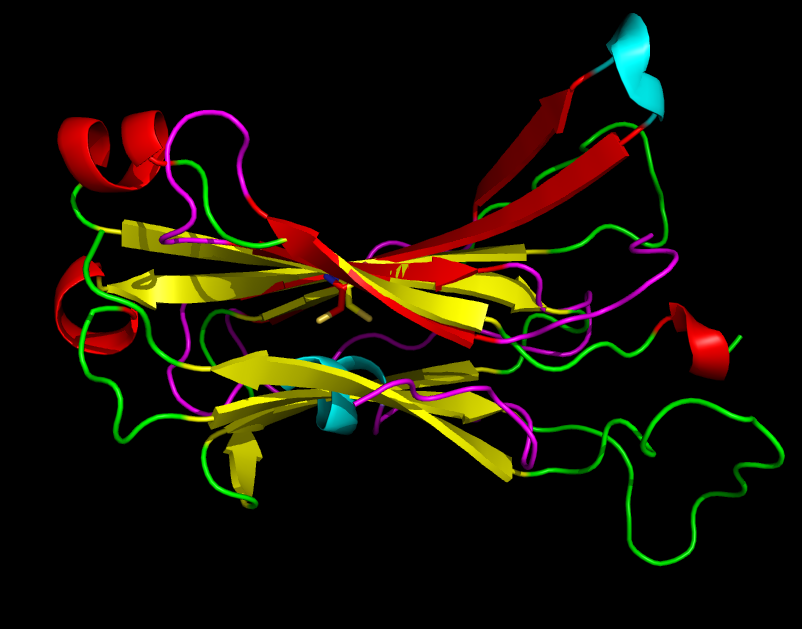
Рисунок 10. Совмещение доменов по консервативным цистеинам. Окраска как на рисунке 7. Совмещаемые цистеины выделены в виде sticks.
В результате такого наложения хорошо совпало 2 β-тяжа (ближние на рисунке 10). Примечательно, что цистеины оказались развёрнуты в разные стороны, не смотря на одинаковый ход цепи. Топология совмещаемых листов одинакова, но остальная укладка доменов сильно разнится в виду разного устройства архитектур, хоть и общий ход полипептидной цепи примерно совпадает. Совмещение доступно в виде pdb-файла.
Также стоит отметить, что полученное совмещение β-листов немного лучше, чем просто совмещение с помощью команды align (см. рис. 11).
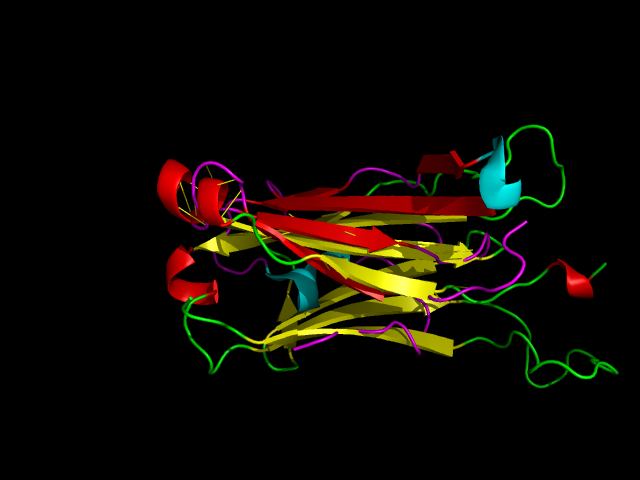
Рисунок 11. Совмещение доменов с помощью align PyMol. Окраска как на рисунке 7.