В директорию /nfs/srv/databases/ngs/potapenko/pr11 были скачаны файл с ридами chr22.fastq и файл с хромосомой chr22.fasta. В этой же директории были запущены команды:
fastqc chr22.fastq java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr22.fastq chr22_clean.fastq TRAILING:20 MINLEN:50 fastqc chr22_clean.fastq
Программа trimmomatic удаляет риды плохого качества. В данном случае она запущена с параметрами TRAILING:20 (отрезает концы ридов, содержащие нуклеотиды с качетством ниже 20) и MINLEN:50 (удаляет риды, длина которых меньше 50 нуклеотидов).
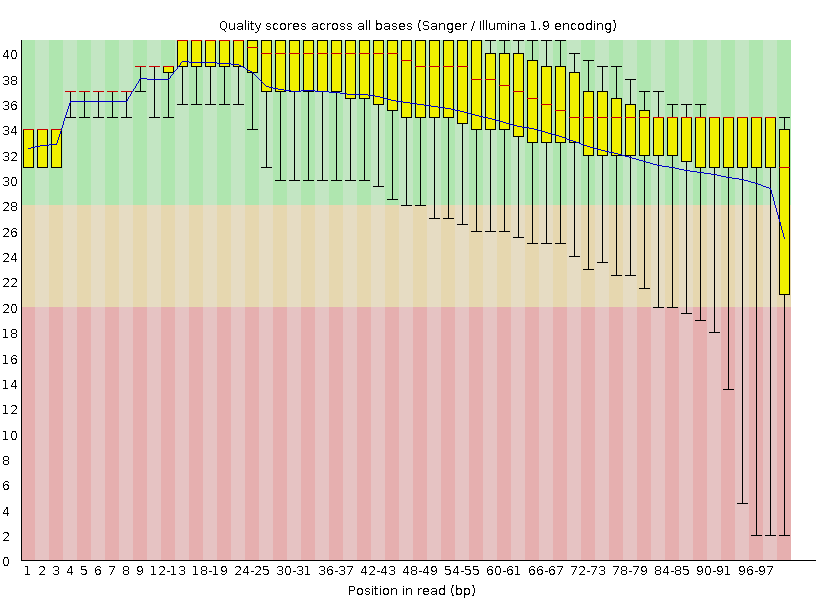
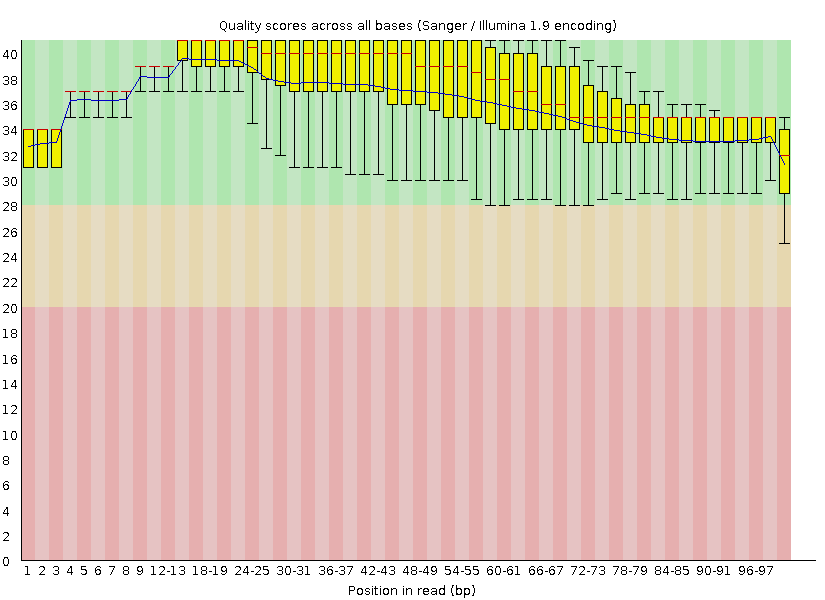
Программа fastqc оценивает качество чтений и выдает анализ в формате html. Графики из анализа изначальных ридов и ридов, обработанных программой trimmomatic, показаны на рисунках 1 и 2. Видно, что после обработки trimmomatic количество ридов уменьшилось (c 11427 до 11091, т.е. были удалены риды, имеющие длину от 30 до 50 нуклеотидов, их 3%), а качество ридов возросло (видно, что плохие концы обрезаны, поэтому качество концов ридов в среднем выше). Выход программы указывает, что изменился GC-состав (с 51% до 50%), но я думаю, что различие даже меньше, чем в процент (из-за округления в разные стороны до целого).
| | |
| Рисунок 1. Анализ качества ридов (файл chr22.fastq). | Рисунок 2. Анализ ридов (файл chr22_clean.fastq). |
Сначала раференсная последовательность хромосомы (chr22.fasta) была проиндексирована программой hisat2-build. В результате работы команды было создано 8 файлов (chr22_index.1.ht - chr22_index.1.ht), обращаться к этой базе нужно по слову chr22_index. Команда запускалась строкой:
hisat2-build chr22.fasta chr22_index
Затем риды (обрезанные и отобранные, файл chr22_clean.fastq) были выровнены на референсную последовательность хромосомы с помощью команды hisat2. Опция -х указывает на проиндексированную базу, -U - на файл с ридами, -S - на выходной файл с выравниванием. Текст команды:
hisat2 --no-spliced-alignment --no-softclip -x chr22_index -U chr22_clean.fastq -S chr22_aligned.sam
В результате работы hisat2 создается файл с выраваниванием (формат .map) и выводится в консоль следующая информация:
11091 reads; of these: 11091 (100.00%) were unpaired; of these: 53 (0.48%) aligned 0 times 11026 (99.41%) aligned exactly 1 time 12 (0.11%) aligned >1 times 99.52% overall alignment rate
Видно, что подавляющее число ридов (99.41%) выровнялось ровно на один участок референса, 53 рида не выровнялись, а 12 ридов выровнялись более 1 раза.
Выравнивание ридов на референс было переведено из формата .sam в формат .bam командой, найденной в примерах руководства
samtools view -bS chr22_aligned.sam > chr22_aligned.bam
Формат .bam бинарный, поэтому читать его "глазами" нельзя, но именно с ним работают многие программы. В том числе это связано с тем, что в этом формате файлы сжаты и занимают меньше памяти (с помощью ls -l можно посомтреть, что файл chr22_aligned.bam занимает на порядок меньше памяти, чем chr22_aligned.sam, хотя, как я понимаю, содержит всю ту же информацию.) Далее порядок ридов был отсортирован в соответствии с координатами участков в геноме, на которые откартировались эти риды. Синтаксис:
samtools sort -T ./tmp/sorted.tmp -o chr22_sorted_alignment.bam -O bam chr22_aligned.bam
Файл с упорядоченными ридами был проиндексирован, в результате чего появился файл chr22_sorted_alignment.bam. (с точкой на конце):
samtools index chr22_sorted_alignment.bam
samtools mpileup -uf chr22.fasta chr22_sorted_alignment.bam -o chr22_SNPs.bfc bcftools call -cv chr22_SNPs.bfc -o chr22_SNPs.vcf
Первой командой был создан файл chr22_SNPs.bfc, в котором записаны отличия ранее выровненных на референс ридов от референса; файлы формата .bfc бинарные (хотя первые первые несколько строк в нем можно прочитать, если открыть файл обычным способом. Строки с заменами не читаются и выглядят как набор смайликов и букв непонятных языков - я так думаю, что это символы, имеющие в кодировке большие номера). Вторая команда переводит формат .bfc в формат .vcf. Этот формат уже "читается глазами", хотя и не очень удобно: на каждой строке указан одна замена, и после нее идет перечисление разных ее характеристик. Не все сокращения интуитивно понятны, все расшифровки даны в начале файла.
Таблица 1. Полиморфизмы (примеры из файла chr22_SNPs.vcf)
| - | Координаты | Референс | Риды | Тип | Глубина покрытия этого места | Качество ридов в этом месте | Комментарии |
| куда смотреть в файле .vcf | POS | REF | ALT | INFO | DP | QUAL | В начале файла все сокращения расшифрованы. Например, DP == Raw read depth |
| пример 1 | 26220983 | gatatatatata | gATatatatatata | INDEL | 3 | 101.432 | "Скользкое место", произошла вставка двух нуклеотидов. |
| пример 2 | 26159289 | G | A | SNP (транзиция) | 19 | 221.999 | Произошла замена, выское качество и глубокое покрытие, можно доверять. |
| пример 3 | 28484784 | G | T | SNP (трансверсия) | 1 | 6.20226 | Эта позиция покрыта только одним ридом, качество низкое, возможно, в этом месте и нет замены. |
Файл с отличиями ридов от референса был переведен в формат .annovar командой:
convert2annovar.pl chr22_SNPs.vcf -format vcf4 -outfile chr22_SNPs.annovar
Формат .annovar содержит обозначение хромосомы, координату по хромосоме, координаты по чтению, какая произошла замена (буквенные обозначения было\стало и het\hom, то есть представлен ли этот snp в гомо- или гетерозиготе.), качество чтения и покрытие в данной позиции.
Аннотировать нужно было только однонуклеотидные замены, поэтому из полученно файла нужно было убрать все строки с инделями. Строки с инделями - это строки инсерций и делеций, т.е. строки, содержащие "-" либо в колонке с ридами, либо в колонке с референсом. Вспомним, что команда grep ищет в файле строки по подстроке, а -v "инвертирует" его работу (инделей в файле оказалось совсем мало, так что можно было бы удалить эти строки и вручную):
cat chr22_SNPs.annovar | grep -v '-' > chr22_SNPs_noindel.annovar
Всего замен и инделей было найдено 225, после удаления инделей осталось 215 строк с заменами (то есть инделей было 10). в предпоследнем столбце файлов .annovar указано качество прочтения, в последнем - покрытие этой позиции. Бросается вглаза, что покрытие часто совсем плохое: встречаются даже замены с покрытием больше 100, но среднее значение покрытия snp равно 10, медианное значение покрытия snp равно 2, медианное значение качества прочтений замен - 11. То есть больше, чем про половину обнаруженных замен нельзя быть уверенными, что эти замены вообще есть у пациента.
Аннотация по базам данных запускалась командами в той же папке /nfs/srv/databases/ngs/potapenko/pr11, поэтому для входного файла указывалсь только его название, а для обращения к базе данных, лежащей в другой папке, указывался полный путь. Аннотация по refgene:
annotate_variation.pl -out refgene -build hg19 chr22_SNPs_noindel.annovar /nfs/srv/databases/annovar/humandb/
В результате выполнения команды были созданы три файла. При этом в файл refgene.log была записана та же информация, что команда выводит на консоль в процессе работы (поля NOTICE, в том числе там записана сама команда и время, когда она была запущена; число обработанных строк и т.д.), в файле refgene.variant_function - сами замены с указанием, в каком экзоне\интроне находится замена. Все мутации разделены на группы: exonic (26), intronic (182), ncRNA_exonic (1), ncRNA_intronic (6). В файле refgene.exonic_variant_function отфильтрованы только мутации в экзонах, для них указано, являются они синонимическими или нет. В этом файле 26 строк, то есть нашлось 26 полиморфизмов, лежащих в экзонах, из них 18 несинонимических (оставшиеся 26 - 18 = 8 синонимические). Следующая аннотация:
annotate_variation.pl -filter -dbtype snp138 -build hg19 -out dbsnp chr22_SNPs_noindel.annovar /nfs/srv/databases/annovar/humandb.old/
Опять созданы три файла, и опять в файл с расширением .log записаны пояснения о работе программы, в файл dbsnp.hg19_snp138_dropped - те замены, которые есть в базе snp138, таких 175 штук. Это те замены, которые имеют идентификатор SNP (обозначается rs + набор цифр). В файл dbsnp.hg19_snp138_filtered отфильтрованы те замены, которых в этой базе нет (и которые, соответственно, не имеют идентификатора rs).
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out genoms1000 chr22_SNPs_noindel.annovar /nfs/srv/databases/annovar/humandb.old/
Созданы три файла, в файле genomes1000.hg19_ALL.sites.2014_10_dropped находятся замены, которые есть в базе 1000genomes (версии 2014 года), их 171. Единственное интересное здесь - в выходной файл добавился столбец (которого не было в других файлах формата .annovar), содержащий числа от 0 до 1. Столбцы не подписаны, но я думаю, что это процент этой замены в популяции (ну не в популяции, а среди геномов в базе данных). Частоты принимают значение от 0 до 1, минимальное значение - примерно 0.3%, среднее - 37% (но эта цифра не очень показательная, т.к. это "средняя температура по больнице" для замен с частотой меньше 10% и больше 90%, что одно и то же), тем не менее, довольно много часто встречающихся замен с частотой в десятках процента.
annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog chr22_SNPs_noindel.annovar /nfs/srv/databases/annovar/humandb.old/
Создано два файла: gwas.log и gwas.hg19_gwasCatalog. Второй файл содержит всего три строки - три замены данного пациента, которые есть в базе gwas Catalog (gwas расшифровывается как genome-wide association study, поэтому в этой базе ассоциации снипов с предрасположенностями к болезням. Веб-версия ebi.ac.uk/gwas ). В файле записываются только названия ассоциированной с полиморфизмом болезни, поэтому более подробную информацию нужно смотреть на сайте. Искать можно либо по названию ассоциированной болезни, либо по rs-идентификатору, посмотрев его по координате в файле chr22_SNPs.annovar. Все три аннотированные замены имеют хорошее покрытие.
annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out clinvar chr22_SNPs_noindel.annovar /nfs/srv/databases/annovar/humandb.old/
Создано три файла. В файле clinvar.hg19_clinvar20150629_dropped всего две строки, в обеих строках стоит ассоциированный диагноз "Focal_segmental_glomerulosclerosis" - патология почечной ткани. Если найти эти же замены в аннотации по refgene, видно, что они лежат в одном локусе (идентификатор NM_001136541, это один из транскриптов липопротеина, взаимодействующего с алипопротеином L1).
Полученные пять аннотаций были сведены в общую таблицу с помощью скрипта.
Стоит отметить, что было взято два файла, полученных аннотацией по refgene, так как они оба содержат полезную информацию, в остальных случаях аннотации-фильтрации брался тот файл *_dropped. Информация, полученная про аннотации по gwas и clinwar, добавлялась вручную (т.к. ее совсем мало). В таблице замены упорядочены так же, как и в исходном файле chr22_SNPs_noindel.annovar - по координате.
Вернуться на страницу семестра
© potapenko 2017-2018