Семестры
Сайт ФББ МГУ
Kodomo Wiki
NCBI
RanHummer personal web-site
Комплексы ДНК-белок
Задание 1. Предсказание вторичной структуры заданной тРНК
Упражнение 1. Предсказание вторичной структуры тРНК путем поиска инвертированных повторов.Вторичную структуру РНК можно предсказать с помощью программы einverted, которая находит инвертированные участки в нуклеотидных последовательностях. Для того, чтобы получить наиболее правдоподобную структуру, можно действовать следующим образом:
-
Подать на вход программе целую последовательность и минимальные параметры, идущие в score с отрицательным знаком (gap_penalty, mismatch_score), также минимизировать параметр minimum score threshold и максимизировать match_score. Тогда получится схема, показывающая, взаимодействуют ли концы структуры. В данном случае это так:
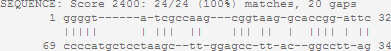
Видно, что в начале и в конце этого "выравнивания" программа нашла спаренные нуклеотиды (блоки по 5 и 4 соответственно). Значит можно ожидать, что в структуре эти стебли действительно есть. В середине же блоков с длиной, большей трех, нет, а также много гэпов, что говорит о недостоверности предсказания этих взаимодействий.
-
Далее нужно подать программе на вход последовательности, стоящие между найденными блоками, и проверить, появятся ли там стебли длины 3-4 и больше, при этом надо увеличить штрафы за несовпадения и за гэпы. Получается:
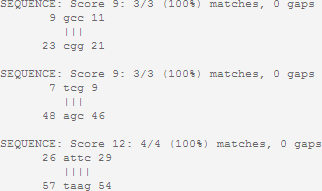
Полученный результат уточняет результат первого прогона программы и приближает к результатам из предыдущего практикума
Далее, была использована программа RNAfold, реализующая алгоритм Зукера. При этом была выбрана не структура с минимальной энергией (рис 3), наиболее похожая на тРНК.
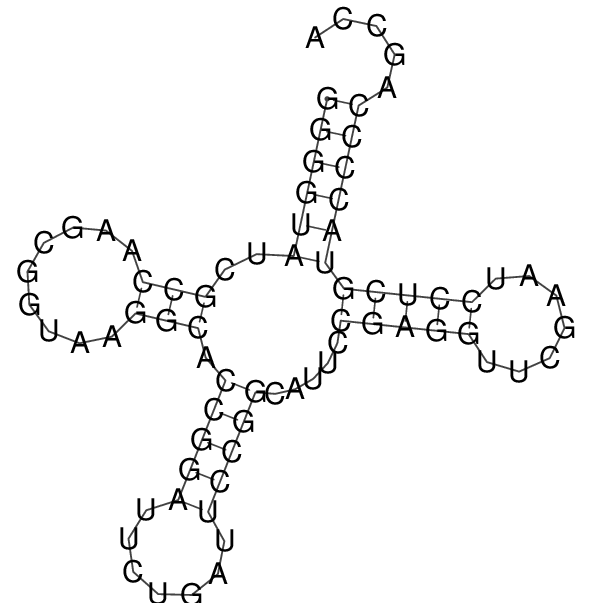
Рисунок 3. Структура с минимальной энергией согласно алгоритму Зукера, наиболее близкая к тРНК.
Результаты, полученные ранее с помощью программы find_pair Вы можете посмотреть здесь. Сравнение предсказаний трех различных программ приведено в таблице 1.
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
|---|---|---|---|
| Акцепторный стебель | 2:7 - 66:71 | 1:5 - 65:69 | 1:6 - 64:69 |
| D-стебель | 10:12 - 23:25 | 9:11 - 21:23 | 9:11 - 21:23 |
| Т-стебель | 49:53 - 61:65 | не выяснено | 47:51 - 59:63 |
| Антикодоновый стебель | 37:44 - 26:33 | 26:29 - 54:57 | 25:29 - 37:41 |
| Общее число канонических пар нуклеотидов | 22 | 12 | 19 |
Можно заметить, что результаты, полученные с помощью разных программ, различаются либо на сдвиг, либо на длину стебля. Т-стебель из-за сильного сдвига в программе einverted вообще не находит себе места.
Задание 1. Поиск ДНК-белковых контактов в заданной структуре.
Упражнение 1. Вспомнить, как с помощью команды define JMol задавать множества атомов.Результат представлен на странице JMol
Упражнение 2. Описать ДНК-белковые контакты в заданной структуре. Сравнить количество контактов разной природы.Для определения количества полярных и неполярных контактов был написан скрипт, определяющий множества полярных и неполярных атомов в белке и в ДНК и считающий, сколько атомов каждого вида располагается рядом (на расстоянии меньше 3.5А для предполагаемых полярных и меньше 4.5 для предполагаемых неполярных контактов). Результаты работы скрипта приведены в таблице 2.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
|---|---|---|---|
| остатками 2'-дезоксирибозы | 4 | 22 | 26 |
| остатками фосфорной кислоты | 11 | 17 | 28 |
| остатками азотистых оснований со стороны большой бороздки | 3 | 5 | 8 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 0 | 0 |
Из таблицы 2 видно, что скрипт нашел больше неполярных контактов, что может быть связано как с их распространенностью, так и с тем, что неполярные контакты описывались в радиусе 4.5А, а полярные - 3.5А. Также видно, что из полярных контактов наиболее представлены контакты с остатками фосорной кислоты. Также не было найдено никаких контактов с остатками азотистых оснований со стороны малой бороздки.
Упражнение 3. Получить популярную схему ДНК-белковых контактов с помощью программы nucplot.С помощью программы nucplot была получена схема ДНК-белковых контактов (рис. 4).
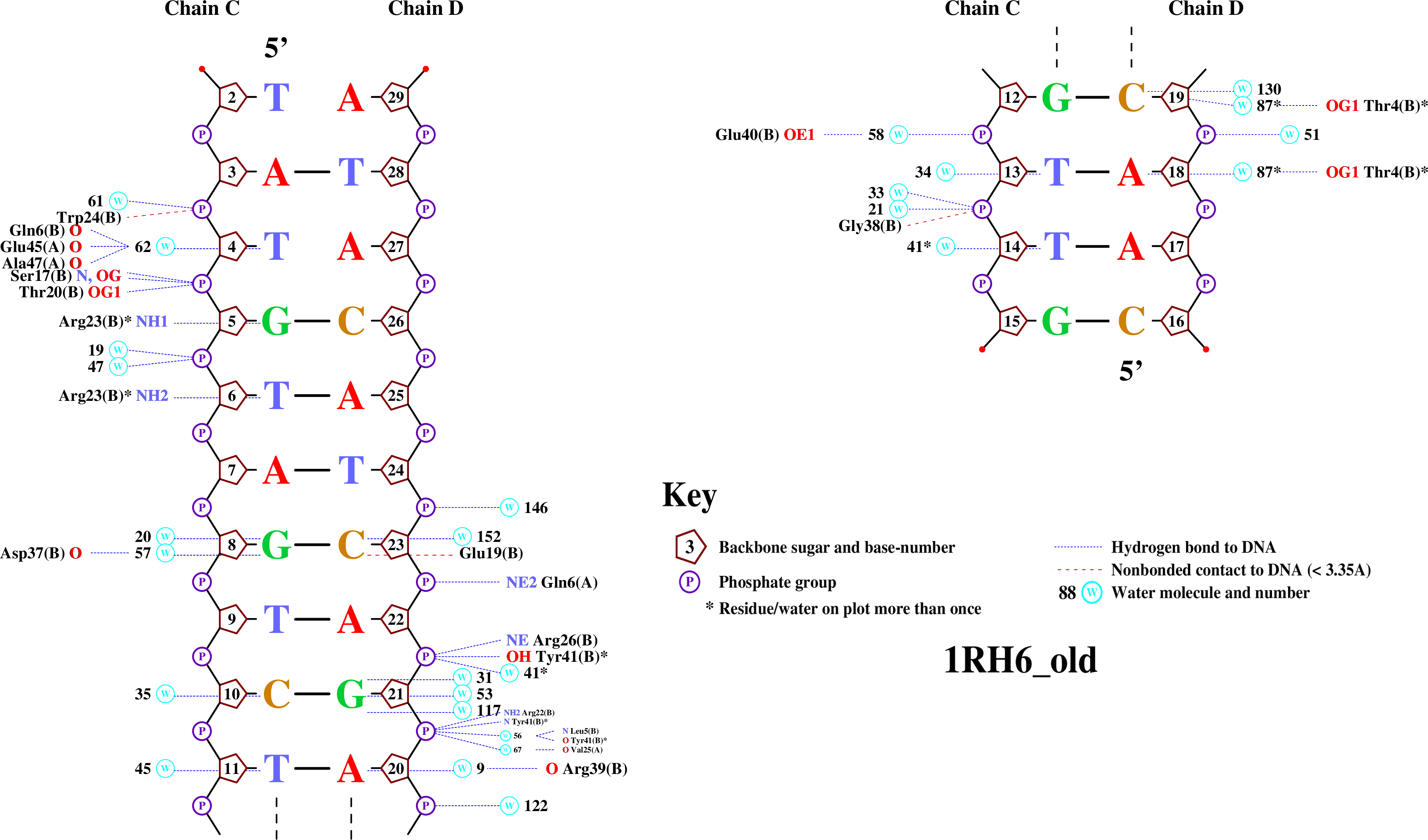
Рисунок 4. Схема ДНК-белковых контактов для цепей С и D, полученная с помощью программы nucplot.
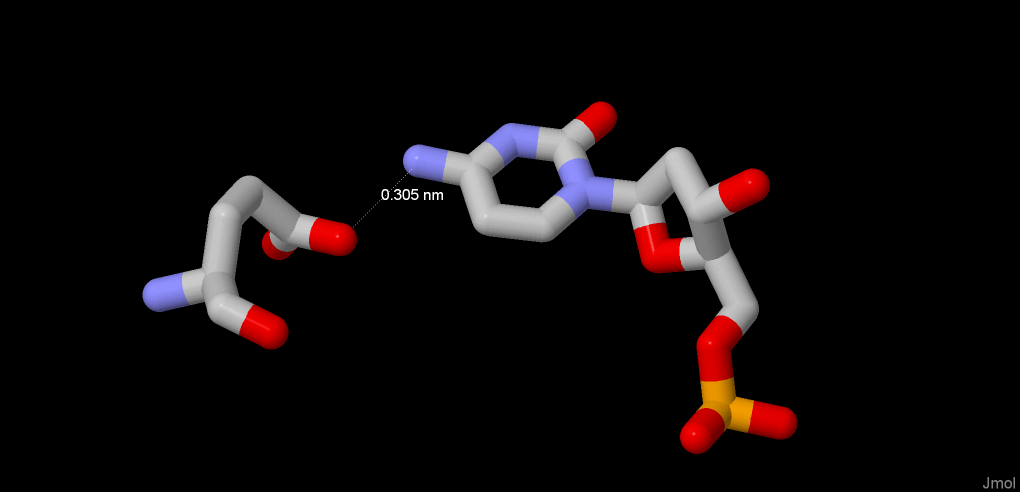
Упражнение 4.Максимальное количество контактов ДНК имеет с Arg23(B) (2 контакта). Они представлены на рис. 5:
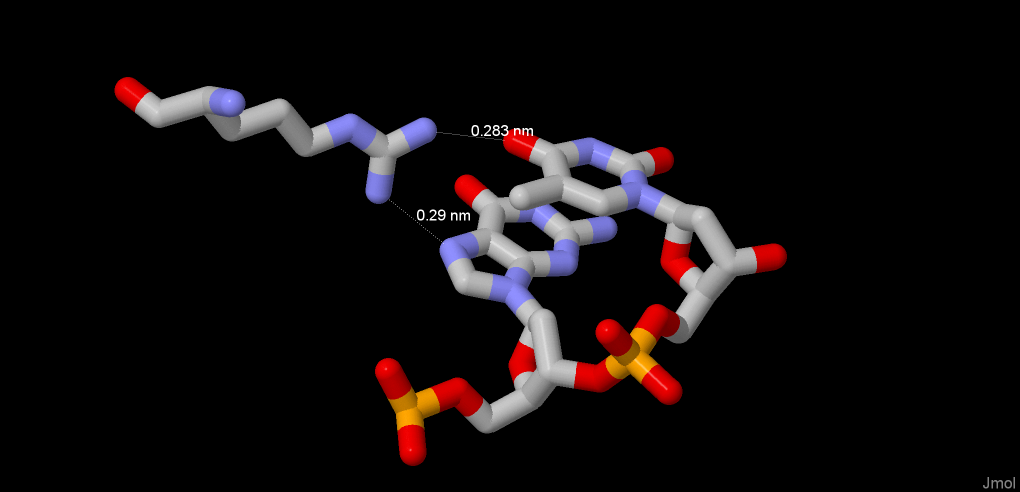
Рисунок 5. Иллюстрация контакта Arg23(B) с 5G и 6T цепи C.
Если говорить о том, какой остаток важен для распознавания фрагмента ДНК, то можем отметить остаток Glu23(B), так как с их участием происходит непосредственное образование водородных связей между остатком азотистого основания и боковым радикалом цепи белка. Это представлено на рис. 6: