Поиск по сходству (нуклеотидный blast)
Задание 1. В первом задании необходимо было определить таксономию и
функцию прочтенной в практикуме 6
последовательности. Последовательность в формате fasta была подана на
вход в blastn. Поиск осуществлялся по базе данных Nucleotide collection со
стандартными параметрами алгоритма megablast (за исключением количества
находок: 1000). Так было построено 225 выравниваний (скачать таблицу в
Excel:
task1.xlsx).
На рис.1 представлена выдача blastn, а на рис.2 представлена суммарная
статистика по всем находкам.
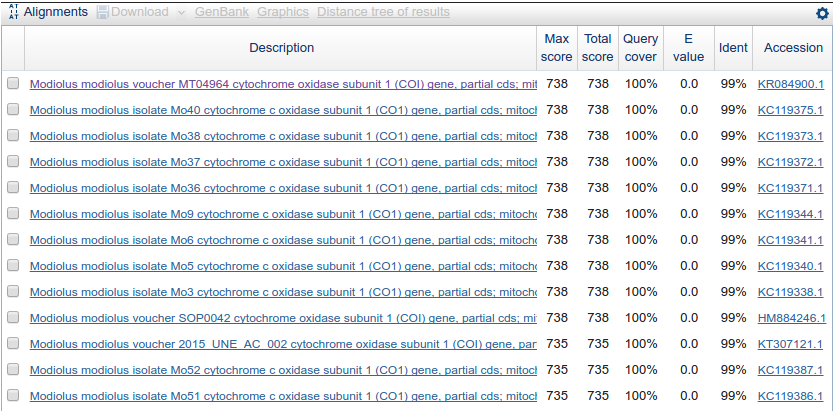 Рис. 1.
Часть найденных последовательностей Рис. 1.
Часть найденных последовательностей |
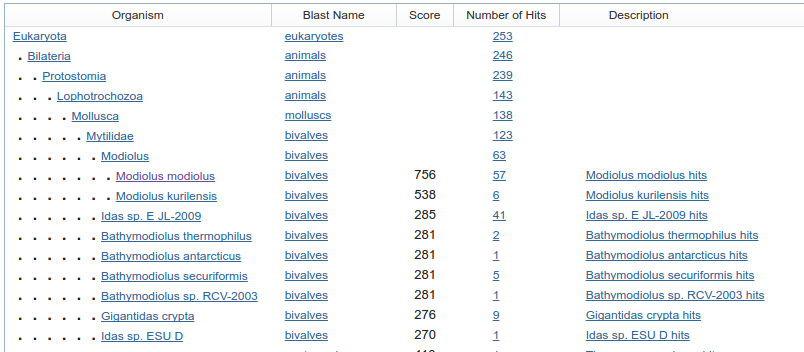 Рис. 2.
Часть из суммарной статистики по всем находкам Рис. 2.
Часть из суммарной статистики по всем находкам |
Первые 42 находки имели следующие характеристики: E-value = 0, Identity = 99%,
Query cover = 100%, Score = [738-726]. Далее шли находки с меньшим покрытием,
идентичностью и весом. Помимо
Modiolus modiolus были найдены
Modiolus kurilensis, M. areolatus, M. comptus, Idas sp. и
Bathymodiolus
manusensis и др. Наибольшего внимания заслуживают первые два организма.
Находки для этих двух видов сильнее всего различаются по score(756 для
M. Modiolus
и 358 для
M. kurilensis) и identity (~99% и ~90% соответственно).
Для наглядности в Jalview было построено выравнивание (Mafft with defaults)
двух последовательностей
этих видов (
M. modiolus 2 сверху) и исходной последовательности (рис. 3).
Рис.3.
Выравнивание интересующих нас последовательностей (окраска по нуклеотидам)
 Рис. 4.
Modiolus modiolus1 Рис. 4.
Modiolus modiolus1 |
Выравнивание демонстрирует как родственность двух видов, так и большее число
идентичных позиций для выравнивания с
M.modiolus.
Таким образом, исследуемая последовательность принадлежит виду
Modiolus
modiolus.
Систематическое положение вида:
Домен: Эукариоты
Царство: Животные
Тип: Моллюски
Класс: Двустворчатые
Отряд: Мидиеобразные
Семейство: Митилиды
Род: Modiolus
Вид: M. modiolus
 Рис. 5.
Цитохромоксидаза2 Рис. 5.
Цитохромоксидаза2 |
Кодируемый белок однозначно определяется результатами: все находки (за
исключением полных митохондриальных геномов) —
гены, кодирующие один и тот же белок. Таким образом, данный митохондриальный
ген кодирует субъединицу 1 цитохромоксидазы (CO1).
Цитохром с-оксида́за
(цитохромоксидаза) — терминальная оксидаза аэробной дыхательной цепи
переноса электронов, которая катализирует перенос электронов с цитохрома с на
кислород с образованием воды. Цитохромоксидаза присутствует во внутренней
мембране митохондрий всех эукариот, а также в клеточной мембране многих аэробных
бактерий.
Три большие субъединицы комплекса (в том числе и CO1), гомологичные
бактериальным, несут на себе все необходимые кофакторы и осуществляют основные
реакции катализа, связанные, в том числе, и с переносом протонов.
Мутации,
затрагивающие ферментативную активность или структуру цитохром с-оксидазы,
приводят к тяжёлым нарушениям метаболизма. Такие нарушения обычно проявляются в
раннем детстве и влияют преимущественно на ткани с высоким потреблением энергии
(мозг, сердце, мышцы). Среди множества митохондриальных
заболеваний, заболевания, связанные с дисфункцией или нарушением сборки
цитохромоксидазы, считаются самыми тяжёлыми
2.
Задание 2. Во втором задании необходимо было сравнить списки находок
нуклеотидной последовательности тремя разными алгоритмами blast. Так как для
заданной мне последовательности не удавалось настроить поиск так, чтобы находок
blastn было хотя бы меньше тысячи, была выбрана другая нуклеотидная
последовательность — ген TK_RS03785, кодирующий
глицеральдегид-3-фосфат дегидрогеназу у археи
Thermococcus kodakarensis,
с которой я уже работала в предыдущих практикумах.
 Рис. 6.
Глицеральдегид-3-фосфат-дегидрогеназа Рис. 6.
Глицеральдегид-3-фосфат-дегидрогеназа |
Глицеральдегид-3-фосфат-дегидрогеназа — фермент массой 37кДа, в
нормальных условиях в цитоплазме находится в форме тетрамера. Данный фермент
катализирует шестую стадию гликолиза (т.е. оксиление глицеральдегид-3-фосфата).
Помимо этого есть данные об участии
ГАФД в неметаболических процессах, включая активацию транскрипции, инициацию
апоптоза и т.д.
3
Таблица 1. Описание параметров худших находок разных
алгоритмов
|
blastn |
megablast |
discontiguous megablast |
| Число находок |
31 |
14 |
29 |
| Max score |
632 |
429 |
632 |
| E-value |
2е-180 |
6е-116 |
2e-180 |
| Identity |
74% |
75% |
74% |
| Query cover |
99% |
99% |
99% |
| Таблица с находками |
blastn.xlsx |
megablast.xlsx |
dmegablact.xlsx |
Для выполнения задания использовались алгоритмы blastn, megablast и discontiguous
megablast. Для уменьшения количества находок (ГАФД, очевидно, есть не только у
T. kodakaraensis) поиск был ограничен семейством Thermococcaceae. В
таблице 1 приведено описание худших находок этих алгоритмов (лучшие находки
идентичны).
По алгоритму blastn было найдено на 2 последовательности больше, чем по
алгоритму discontiguous megablast; однако обе эти находки имеют очень высокий
e-value и очень низкое покрытие (рис 7).
 Рис. 7.
Две находки, найденные только с помощью blastn Рис. 7.
Две находки, найденные только с помощью blastn |
Выводы (в квадратных скобках указаны значения по умолчанию):
- алгоритм blastn предназначен для поиска не обязательно
родственных, но похожих последовательностей. Поэтому среди находок
могут быть и негомологичные последовательности, которые не нужно
учитывать при дальнейшем анализе. Кроме того, поиск довольно медленный,
но зато размер слова* задается в пределах от 7 до 15 [11];
- алгоритм discontiguous megablast предназначен для межвидового
поиска гомологов. При этом в слове* допускаются мисматчи, а его
размер задается в пределах от 11 до 12 [11];
- алгоритм megablast предназначен для поиска близкородственных
последовательностей и особенно подходит для поиска на 95% идентичных
находок. Помимо этого этот алгоритм самый быстрый. Однако размер
слова* можно задать только в пределах от 16 до 256 [28].
*
Слово, инициирующее выравнивание (initial seed) — слово
определенной длины, с которого начинается выравнивание. BLAST ищет совпадения
слов определенной длины между входной
последовательностью и последовательностями из банка; найденное слово инициирует
дальнейшее полное выравнивание. При этом мисматчи либо допускаются, либо нет в
зависимости от алгоритма.
Задание 3. В этом задании нужно было проверить наличие гомологов трех
белков в заданном геноме
Amoeboaphelidium protococarum. Этот организм
относится к афелидам, т.е. таксону родственных грибам протистов из группы
опистоконт. Все описанные виды являются внутриклеточными паразитами. Афелиды
обладают классическим набором органелл эукариотической клетки
4.
В качестве белков были выбраны:
- CISY_HUMAN — митохондриальная цитратсинтаза;
- HSP7C_HUMAN — консервативный шаперон HSP70, белок теплового
шока; имеется у большинства организмов из всех царств;
- PABP2_HUMAN — белок, связывающий поли(А) хвост матричной РНК.
Для выполнения задания использовался локальный tblastn. С его помощью был
осуществлен поиск гомолога белка в "трансляции" нуклеотидного банка (наш геном).
Таким образом, для каждого белка было получено 3 файла с построенными
выравниваниями.
1) CIZY-HUMAN (цитратсинтаза) — фермент, катализирующий реакцию
конденсации ацетата (ацетил-CoA) и оксалоацетата, в результате чего образуется
цитрат. Синтетаза цитрата обнаружена практически во всех клетках аэробных
организмов, катализируемая реакция является лимитирующей на первом этапе цикла
трикарбоновых кислот. Фермент располагается в митохондриальном матриксе эукариот,
однако кодируется ядерным геномом. Синтез осуществляется на рибосомах цитоплазмы,
а затем синтаза цитрата транспортируется в матрикс митохондрии
5.
Можно предположить, что такой белок должен быть у всех аэробных
организмов.
Открыть файл с выравниваниями:
cizy.fasta.
Всего было построено 4 выравнивания, из которых стоят внимания только два
идентичных выравнивания для двух разных скэффолдов: scaffold-693 и scaffold-157.
Характеристика выравниваний:
- score: 565 (564)
- e-value: 2(5)e-180
- identities:69% из них positives:84%
Значение параметра e-value отличное и к тому же 69% идентичных нуклеотидов,
позволяют предположить, что у исследуемого организма действительно имеется
гомолог цитратсинтазы.
2) HSP7C_HUMAN — белок-шаперон из семейства белков теплового шока
массой 70 кДа (heat shock 70 kDa proteins — Hsp70). С-конец включает домен,
связывающий субстрат, а N-конец — АТФ-связывающий домен. Между двумя
доменами находится консервативный участок, называемый петлей LL,1. Ближе к
С-концу, возможно, располагается сайт для докинга ко-шаперонов. При связывании
с полипептидами способстует их правильному фолдингу. Также принимает участие в
апоптозе, росте и дифференциации клеток, сигнальной трансдукции
6,7.
Открыть файл с выравниваниями:
hsp7c.fasta.
Всего blast построил 16 выравниваний. Лучшее из них имеет параметры:
- > scaffold-199
- score: 917
- e-value: 0.0
- identities: 78% из них positives:89%
Мне кажется, что такие данные позволяют предположить, что гомолог и этого белка
тоже присутствует у исследуемого организма. Хочется также заметить, что blast
построил несколько выравниваний, в которые вошли лишь некоторые участки нашего
белка. Например, одно из таких выравниваний имеет следующие параметры:
- > unplaced-999
- score: 549
- e-value: 5e-174
- identities: 83% из них positives: 92%
Так как 1-386 а.о. входят в состав АТФ-связывающего домена
7, можно
сделать вывод, что у исследуемого организма есть гомологичный домен. Кроме того,
этот участок может быть частью более крупного гомологичного участка, который в
данный момент разбит на части, так как точное расположение unplaced-999 в
геноме нашего организма пока неизвестно.
3) PABP2_HUMAN — полиаденилат-связывающий белок 2. Этот белок
участвует в присоединении поли(А)-хвоста к 3'-концу пре-МРНК. Также присутствует
на разных стадиях метаболизма мРНК
8. Полиаденилирование защищает мРНК
от ферментативного разрушения в цитоплазме, способствует терминации транскрипции, участвует в экспорте мРНК из ядра и трансляции. Практически все эукариотические
мРНК полиаденилируются
9.
Открыть файл с выравниванием:
pabp2.fasta.
Всего было построено 18 выравниваний. Все выравнивания, кроме двух, не имеют
никакой статистической ценности. Обе находки идентичны одному и тому же участку
168-253 а.о. Параметры:
- > scaffold-100, > scaffold-199
- score: 117 (114)
- e-value: 2e-28 (2e-27)
- identities: 63% из них positives: 74% (73%)
Согласно UniProt участок с 155 по 306 а.о. важен для гомоолигомеризации.
Возможно, есть гомология доменов, хотя мне это
кажется маловероятным из-за маленького числа идентичных позиций
для такого небольшого участка.
Задание 4. В четвертом задании было необходимо найти один ген белка,
закодированный в одном скэффолде
A. protococarum. С помощью команды
(1) из файла
X5.fasta была получена информация о длинах
скэффолдов (
scaffolfs.out).
Затем был выбран скэффолд №6
длиной в пару десятков тысяч пар нуклеотидов. С помощью команды (2) была получена
последовательность данного скэффолда (
scaffold6.fasta
).
(1) infoseq X5.fasta -only -name -length > contigs.out
(2) seqret X5.fasta:scaffold-6 -out scaffold6.fasta
Затем с помощью алгоритма megablast был осуществлен поиск наиболее
правдоподобного гена, закодированного вданном скэффолде. Так как интроны у
A. protococarum короткие, ген может поместиться в одном таком скэффолде.
Так как для данного рода помимо
A. protococarum нет ни одного
представителя с секвенированным геномом, поиск по роду не дал бы результатов.
Поэтому поиск blast был
ограничен по Opisthokonta и Fungi, так как наш вид принадлежит группе
опистоконт и в то же время является родственником грибов. На рис.8 и 9
представлена часть находок.
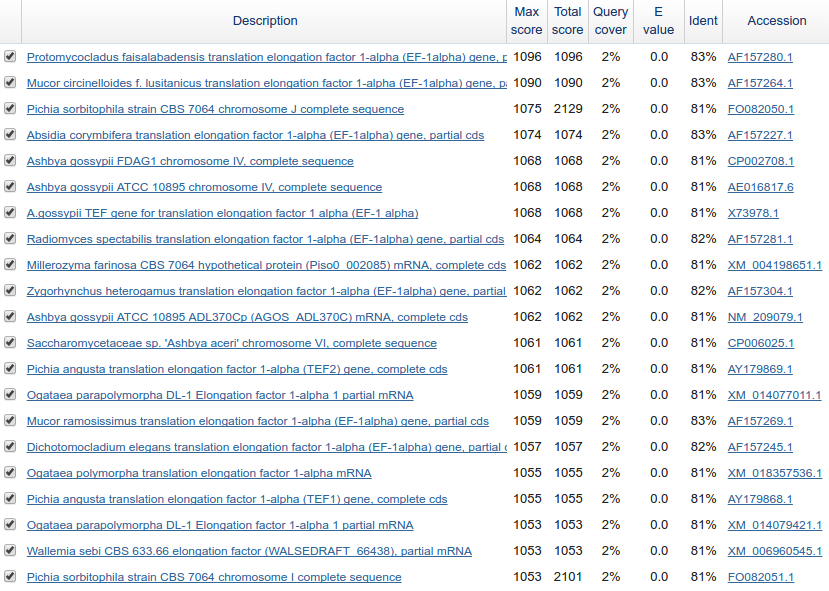 Рис. 8.
Часть находок (ограничение по грибам) Рис. 8.
Часть находок (ограничение по грибам) |
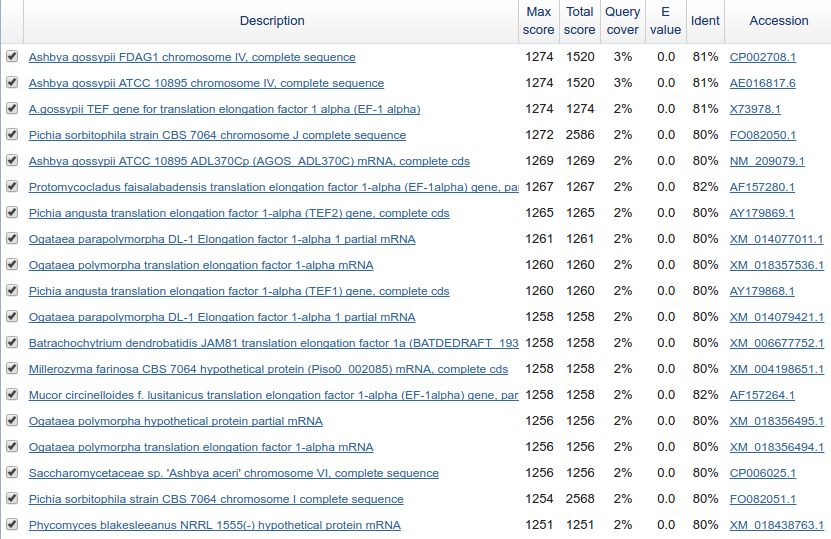 Рис. 9.
Часть находок (ограничение по опистоконтам) Рис. 9.
Часть находок (ограничение по опистоконтам) |
Значение e-value, identity и то, что все найденные гены кодируют фактор
элонгации трансляции 1-альфа, позволяет предположить, что именно этот ген
закодирован в данном скэффолде. На рис. 10 показано примерное расположение
гена в скэффолде (точные границы blast найти не позволяет).

Рис. 10. Примерное
расположение гена |
Факторы элонгации трансляции — регуляторные белки,
взаимодействующие с рибосомами и обеспечивающие процесс элонгации
трансляции
12.
Фактор элонгации трансляции 1-альфа (EF1a) — один из двух факторов
элонгации трансляции у эукариот, осуществляющий доставку аминоацил-тРНК в
аминоацил-сайт рибосомы. Содержится в клетках эукариот в большом количестве
10,11.
Также может обеспечивать один из механизмов внутриклеточного транспорта мРНК
благодаря способности связываться с компонентами цитоскелета
12.
Задание 5. В этом задании нужно было сравнить геномы родственных вирусов,
основываясь на сходстве последовательностей. Для этого задания было выбрано
шесть полных геномов вирусов из рода
Atadenovirus:
- Bovine adenovirus 6 strain 671130 (NCBI)
- Ovine adenovirus 7 (NCBI)
- Psittacine adenovirus 3 isolate HKU/Parrot19 (NCBI)
- Lizard adenovirus 2 isolate 23-06 (NCBI)
- Snake adenovirus (NCBI)
- Bovine adenovirus D (NCBI)
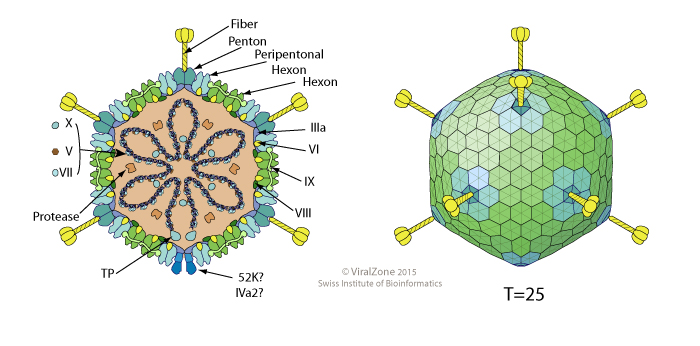
Рис. 11.
Модель атаденовируса13 |
Рис. 12. Микрофотография типичного
аденовируса14 |
Систематика:
Домен: Вирусы
Семейство: Аденовирусы (Adenoviridae)
Род: Атаденовирус (Atadenovirus)
Род
Atadenovirus принадлежит семейству Adenoviridae. Хозяевами являются
позвоночные
15. Вирион (рис. 11) имеет капсид (диаметр = 90нм) в форме
икосаэдра с псевдо Т=25 симметрией без оболочки. Капсид в свою очередь состоит
из 720 субъединиц.
Геном представлен несегментированной линейной двуцепочечной ДНК длиной в 30 кб
и кодирует около 30 белков. Транскрипция состоит из двух этапов: раннего
(репликация) и позднего (сборка вириона). Все гены транскрибируются
РНК-полимеразой II хозяина. При транскрипции из одних и тех же генов могут
образовываться разные мРНК благодаря альтернативному сплайсингу и использованию
разных поли(А)-сайтов
13.
Для выполнения задания все геномы были сохранены в одном файле (
databank.fasta). Затем использовался локально
установленный пакет blastp+, а именно алгоритм tblastx (трансляции входных
последовательностей против трансляций нуклеотидной базы данных). Перед этим
из fasta-файла была создана база данных с помощью команды (1). Затем этот файл
был подан на вход tblastx с табличной выдачей с помощью команды (2). Таким
образом, трансляции каждого генома мы сравнили с трансляциями всех геномов.
После этого из полученного файла (
task5.fasta) с
помощью скрипта
pr8.py были удалены неинформативные и слабо
сходные находки (3).
(1) makeblastdb -in databank.fasta -dbtype nucl
(2) tblastx -query databank.fasta -db databank.fasta -outfmt 7 > task5.fasta
(3) python pr8.py -i task5.fasta -o task5.out -e 0.001 -s 70
Полученный файл был отредактирован в Excel (
task5.xlsx).
Согласно полученной таблице, для геномов
Bovine adenovirus 6
и
Bovine adenovirus D было найдено наибольшее число сходных
последовательностей (190) с наибольшим
максимальным процентом сходства (100%) и относительно высоким средним процентом
сходства (76.69%). Можно предположить, что эти два вируса самые близкие
родственники среди нашей выборки.
Однако дать однозначный ответ, основываясь только на этих параметрах, трудно.
Например, для
Bovine adenovirus 6 и
Ovine adenovirus 7 средний
процент сходства выше (77.78%), в то время как общее число находок (42) и
максимальный процент сходства (88.8%) ниже. В случае сравнения геномов
Snake adenovirus и
Lizard adenovirus 2 максимальный процент
сходства (90%) и общее число находок (76) выше, чем у предыдущих двух геномов,
в то время как средний процент сходства ниже (76,44%). Какой же параметр важнее?
Так как нет единого мнения о том, как описывать филогению вирусов по
последовательностям геномов из-за их быстрой эволюции и частых потерь и
приобретений новых генов, непонятно, как изучать сходство двух геномов вирусов.
Как мне кажется, помимо уже описанных выше способов,
необходимо также учитывать какой процент найденные сходные участки
составляют от всего генома.
Для геномов
Bovine adenovirus 6 и
Bovine adenovirus D была посчитана общая длина найденных сходных участков
(см. лист calculations в таблице), и их процент от длины генома
Bovine
adenovirus D составил 82,6%, что подтверждает родство двух вирусов. В то же
время для
Snake adenovirus и
Lizard adenovirus 2 этот параметр
составил 41,9%, а для
Bovine adenovirus 6 и
Ovine adenovirus 7
— всего лишь 29,6% (вычисления аналогичные, в таблице не приведены),
что говорит о том, что геномы этих вирусов не сильно похожи.
В общем, можно сделать вывод, что геномы всех вирусов из выборки, кроме
Bovine adenovirus 6 и
Bovine adenovirus D, недостаточно
похожи, чтобы говорить об их родстве.

