Мною была выбрана мнемоника RPOB - бета субъединица РНК полимеразы, часть транскрипционного комплекса.
Всего с такой мнемоникой 735 белков.
Идентификаторы выбранных мной белков: P0A8V2, P37870, P9WGY9, P60281, O52271, A0QL49, A1KGE7, A4SHU9, A5GNH3, A5UH20.
Ссылка на выравнивание
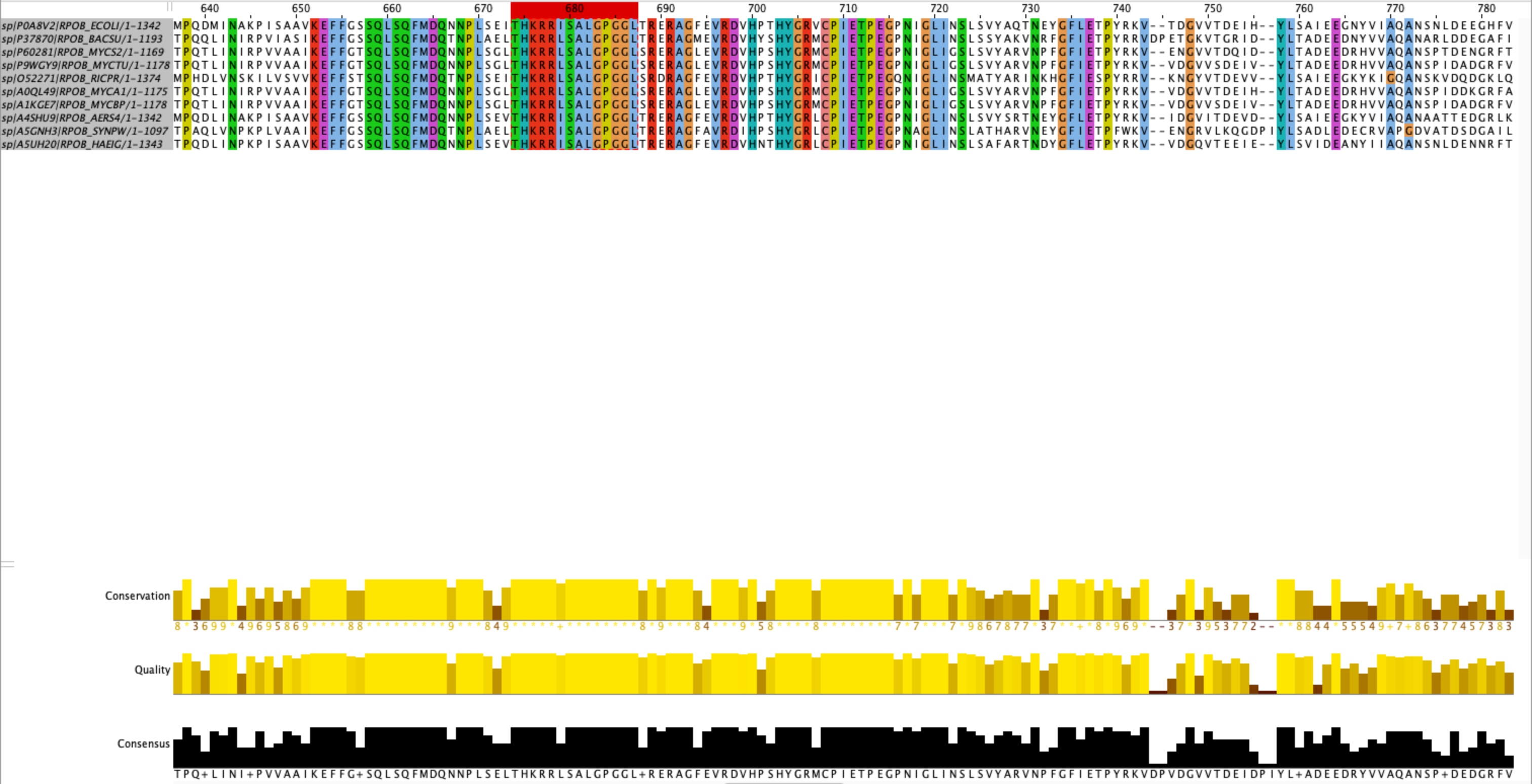
Рисунок 1. Выбранный мной участок для паттерна, позиции выравнивания - 674 - 687.
Из участка выше составляется следующий паттерн: T-H-K-R-R-[LI]-S-A-L-G-P-G-G-L
В итоге при помощи программы fuzzpro, а именно команды:
Был осуществлен поиск по заданному патерну среди всех белков бактерий. Число находок указано в конце файла вывода,
но для удобства нахождения правильно найденных, ложноположительных находок и ложноотрицательных результатов,
я воспользовался функциями bash:
grep "HitCount: 1" out | wc -l
Результат - всего 458 находок по данному паттерну.
Всего белков с моей мнемоникой - 735. То есть не было найдено 284 белка по данному паттерну.
Я также попробовал улучшить паттерн: [TS]-H-[KR]-[KR]-R-[LI]-S-A-L-G-P-G-G-L
Сделал я это на основании того, что серин и треонин близкие по свойствам остатки,
поэтому позицию можно ослабить; лизин и аргинин оба положительно заряжены,
поэтому в этой позиции их можно считать взаимозаменяемыми; аргинин и лизин близки
по химическим свойствам, поэтому в теории мотив сохраняется при замене одного на другой.
Получилось, что всего находок по паттерну - 496, с верной мнемоникой - 489. То есть
получилось верно найти на 38 белков больше, чем по более строгому паттерну.
2. Поиск мотивов в белках программой MEME и поиск этих мотивов в банке
Был произведен поиск мотивов по выбранным мною белкам. Было найдено три мотива:
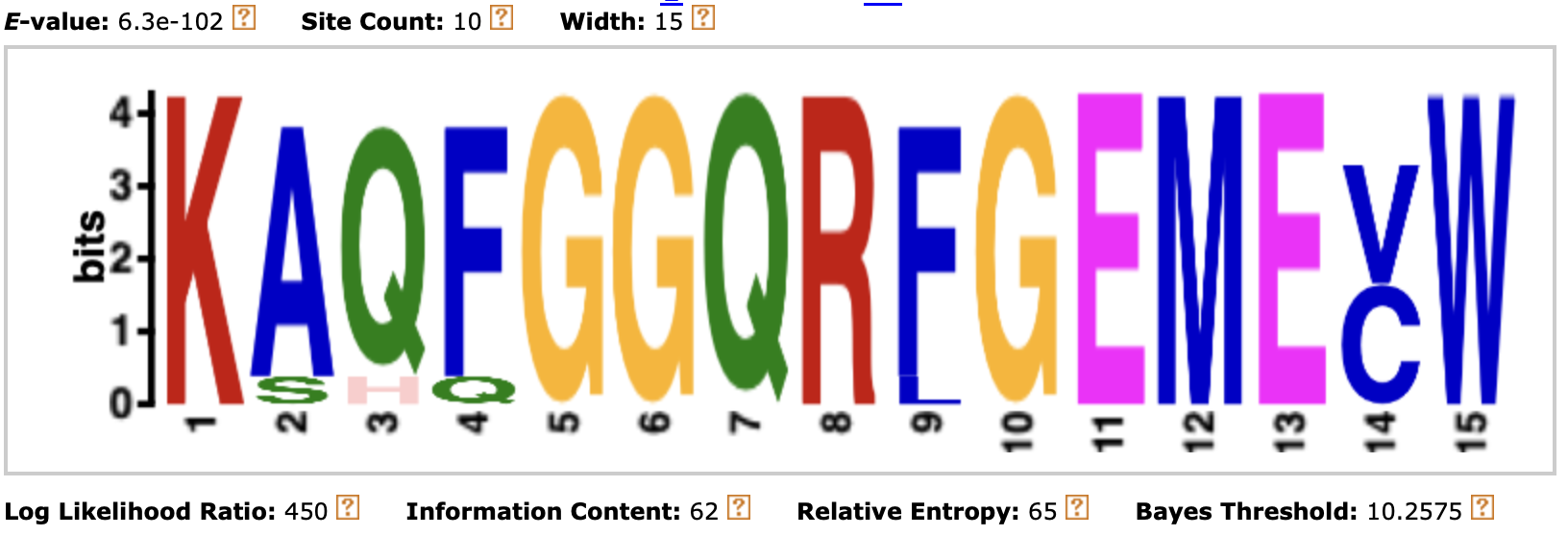
Рисунок 2. Первый найденный мотив при помощи программы meme для моих белков
Рисунок 3. Второй найденный мотив при помощи программы meme для моих белков
Рисунок 4. Третий найденный мотив при помощи программы meme для моих белков
Длина всех мотитов составляет 15 аминокислот. Первый и второй мотив располагаются примерно в центре последовательности
на расстоянии 46 аминокислот друг от друга,
последний мотив располагается ближе к концу последовательности. Достоверности находок подтверждаются низкими e-value и p-value.
Все три мотива были найдены во всех выбранных мною белках.
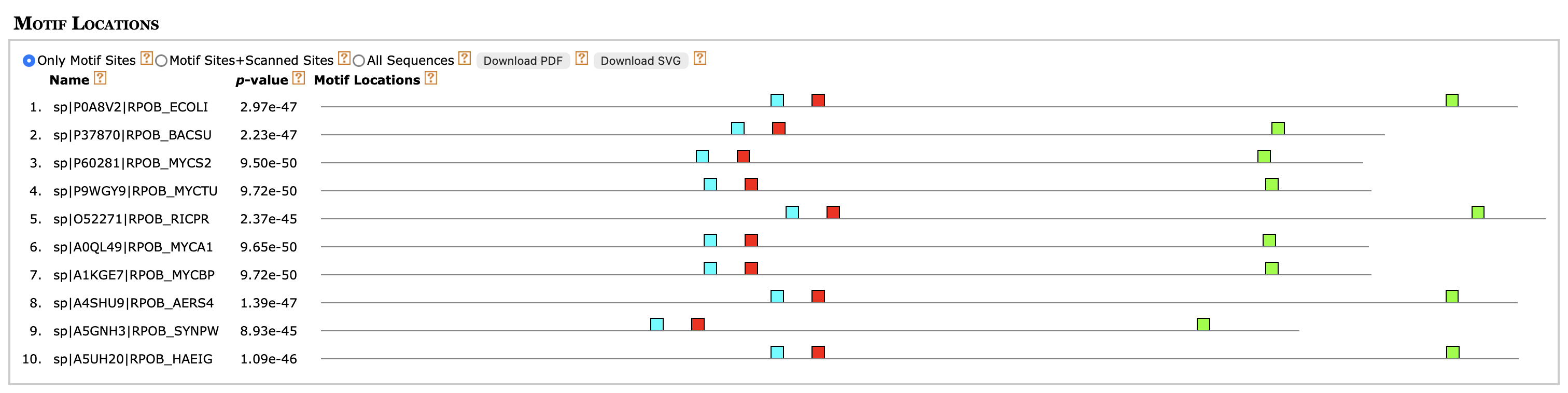
Рисунок 5. Расположение мотивов в моих белках, найденных программой meme
Было найдено 758 белков с мотивами, найденными meme. Почти во всех находках содержались все три мотива, кроме 11-ти последних, тем более, они относятся к другой мнемонике, а также в выдаче дополнительно присутствуют белки с мнемоникой RPOBC, видимо, родственные RPOB. Найденных белков при помощи mast больше, чем белков с данной
мнемоникой в целом, то есть имеется 23 ложноположительных находок. Тем не менее, mast является более чувствительным подходом
по сравнению с fuzzpro.
3. Поиск последовательности Шайна — Дальгарно в геноме своего прокариота
В качестве прокариота я выбрал архею из 1 семестра - Halanaeroarchaeum sulfurireducens. Геномную последовательность и
геномную таблицу в формате gff я скачал из NCBI, использовав референсный геном штамма HSR2. При помощи программы
fazznuc:
fuzznuc -sequence GCF_001011115.1_ASM101111v1_genomic.fna -pattern "AGGAGG" -outfile pshd_forward.out -complement N
fuzznuc -sequence GCF_001011115.1_ASM101111v1_genomic.fna -pattern "AGGAGG" -outfile pshd_complement.out -complement Y
произвел поиск по прямой и комплементарной цепям. В первом случае находок было 1236, во втором - 2371. Чтобы проверить,
имеется ли достоверное отличие числа находок от ожидаемого по случайным причинам, был написан небольшой скрипт на python
для подсчета долей букв в геноме:
from pathlib import Path
from collections import Counter
fasta = Path("GCF_001011115.1_ASM101111v1_genomic.fna")
count = Counter()
total = 0
with fasta.open() as f:
for line in f:
if line.startswith(">"):
continue
for ch in line.strip().upper():
if ch in "ACGT":
count[ch] += 1
total += 1
print("A", count["A"], count["A"] / total)
print("C", count["C"], count["C"] / total)
print("G", count["G"], count["G"] / total)
print("T", count["T"], count["T"] / total)
print("TOTAL", total)
Соответственно, чтобы посчитать вероятность появления мотива, воспользовались формулой P = pA × pG × pG × pA × pG × pG,
и эта вероятность составила 0,00033339. Затем умножаем эту вероятность на длину генома, 2209738 × 0,00033339 × 2 = 1437 ожидаемых
находок. Наблюдаемое число находок (3607) существенно превышает ожидаемое по случайной модели (1473),
рассчитанной по частотам нуклеотидов в геноме. Следовательно, различие достоверно,
а найденный мотив не является случайным.
Далее, используя геномную таблицу, и скрипт на python:
#!/usr/bin/env python3
from pathlib import Path
GFF = Path("genomic.gff")
FWD = Path("pshd_forward.out")
REV = Path("pshd_complement.out")
MIN_UP = 3
MAX_UP = 15
def read_cds(gff_path):
cds = []
with gff_path.open() as f:
for line in f:
if not line.strip() or line.startswith("#"):
continue
cols = line.rstrip("\n").split("\t")
if len(cols) < 9:
continue
if cols[2] != "CDS":
continue
seqid = cols[0]
start = int(cols[3])
end = int(cols[4])
strand = cols[6]
cds.append((seqid, start, end, strand))
return cds
def read_hits(report_path, strand):
hits = []
seqid = None
with report_path.open() as f:
for line in f:
line = line.rstrip("\n")
if line.startswith("# Sequence:"):
seqid = line.split()[2]
continue
parts = line.split()
if seqid and len(parts) >= 2 and parts[0].isdigit() and parts[1].isdigit():
start = int(parts[0])
end = int(parts[1])
hits.append((seqid, start, end, strand))
return hits
def is_correct(hit, cds_list):
hseq, hs, he, hstrand = hit
for cseq, cs, ce, cstrand in cds_list:
if hseq != cseq or hstrand != cstrand:
continue
if cstrand == "+":
dist = cs - he
if MIN_UP <= dist <= MAX_UP:
return True
else:
dist = hs - ce
if MIN_UP <= dist <= MAX_UP:
return True
return False
cds = read_cds(GFF)
hits = read_hits(FWD, "+") + read_hits(REV, "-")
total = len(hits)
correct = sum(1 for h in hits if is_correct(h, cds))
print(f"Всего находок: {total}")
print(f"Правильных: {correct}")
print(f"Процент правильных: {100 * correct / total:.2f}%")
было посчитано общее количество находок, правильных находок относительно старт-кодонов CDS и, соответственно, процент правильных.
Числа составили 3607, 36 и 1.00% соответственно.