Домены и профили
Задание 1: Cписок белков Uniprot с заданным составом доменов Pfam
Для выполнения практикума я выбрала домен Aminotran_5 Pfam и доменную архитектуру, состоящую из этого домена и Fe-S metabolism associated domain. Данные домены представлены у бактерий
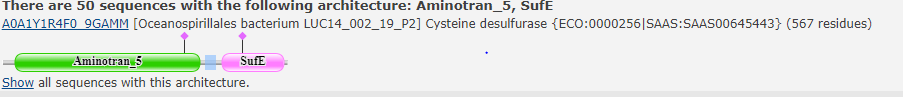
Выбранная доменная архитектура
По данным, предоставленным в PFAM, эта архитектура встречается в 50 белках любых организмов. Однако по данным Uniprot в 147 белках среди бактерий. Возможно база данных PFAM давно не обновлялась.
| ID | AC | Название | Количество белков бактерий, в состав которых входит домен |
| Aminotran_5 | PF00266 | Aminotransferase class-V | 184,872 |
| SufE | PF02657 | Fe-S metabolism associated domain | 16,504 |
Чтобы получить список бактериальных белков, в состав которых входит выбранная доменная архитектура, в расширенный поиск Uniprot было введено: database:(type:pfam pf00266) database:(type:pfam pf02657) taxonomy:bacteria
Таблица с информацией о всех бактериальных белках с выбранной доменной архитектурой
Чтобы определить типиные длины белков, была построена гистограмма длин белков (картинка ниже). Мода длин белков составляет 562.
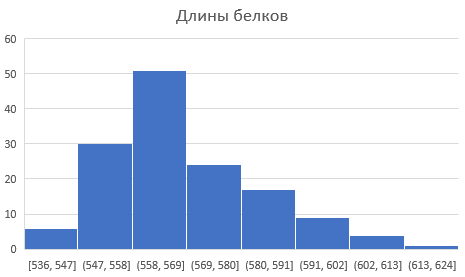
Далее в таблице с информацией о всех бактериальных белках с выбранной доменной архитектурой я оставила только белки с характерной длиной, то есть с длиной из диапазона 558-569. Я решила выбрать такой диапазон, опираясь на полученную гистограмму. Таким образом, мы стремимся получить список белков с нужной архитектурой. Ведь слишком короткие, вероятно, будут содержать только фрагменты доменов, а слишком длинные могут содержать домены, еще не охарактеризованные в Pfam. Всего было выбрано 53 белка
Задание 2: Построение hmm профиля семейства белков с выбранной архитектурой. Проверка его работы.
Получение множественного выравнивания отобранных белков.
Были скачаны отобранные последовательности белков, а затем выровняны командой muscle. Чтобы провести "ревизию" выравнивания я:1) Нашла N-концевой консервативный блок и удалила все колонки до него. 2)Удалила те последовательности, в которых N-концевой консервативный блок отсутствует. 3)Нашла С-концевой консервативный блок и удалила все колонки после него. 4)Также я удалила из множественного выравнивания последовательности, в которых наблюдаются длинные вставки. Таким образом, я удалила 10 последовательностей.
Команды для построения и калибровки HMM профиль выравнивания:
Проверка работы hmm профиля.
Для того, чтобы проверить работу профиля, буду искать белки с заданной архитектурой среди белков, включающих один домен из архитектуры, используя откалиброванный профиль. Из Uniprot я скачала все бактериальные последовательности, содержащие Fe-S metabolism associated домен в fasta формате. Таких последовательнойстей оказалось 17700, в то время как белков с доменом Aminotransferase class-V было найдено 211790. Для поиска белков с заданной доменной архитектурой с помощью hmm профиля использовалась команда:
Сравнение списка находок с исходной таблицей.
Были обнаружены все белки с нужной доменной архитектурой, кроме двух (в дальнейшем я учту это при вычислении чувствительности и мощности). При этом было найдено 3 лишних белка (в таблице они отмечены красным)
Подбор порога веса для предсказания того, что находка имеет нужную доменную архитектуру.
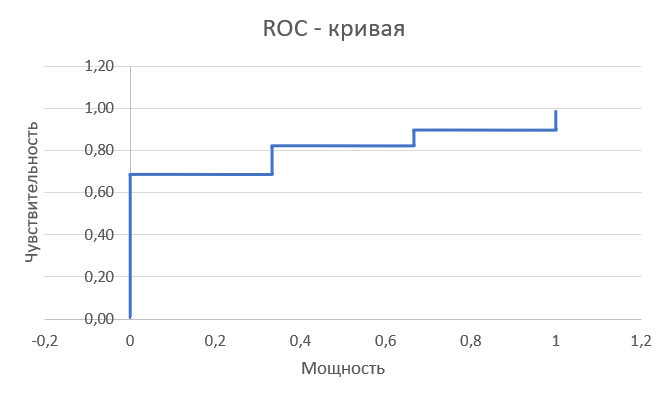
Чтобы подобрать оптимальный порог веса для поиска определенной доменной архитектуры с помощью hmm профиля, я посчитала чувствительность (ось y) и мощность (ось x) теста для разных порогов веса. По этим данным построла ROC - кривую:
Далее я построила распределение весов находок :
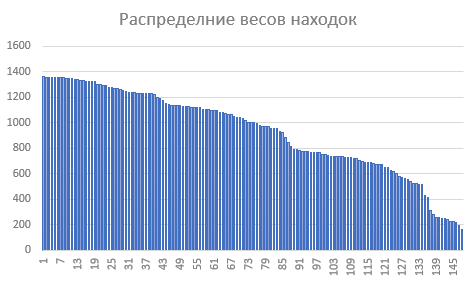
По моим данным наибольшее значение параметра F1 достигается при пороговом значении веса, равном 0. То есть при том, который был выбран с самого начала. Это логично, ведь высталяя такой порог, мы получаем список белков почти полностью соответствующий реальным данным. Содержательно,этот параметр позволяет найти порог, при котором наиболее сбалансированы частоты ложно положительных и ложно отрицательных предсказаний.