Навигация по сайту:
|
Ресеквенирование. Поиск полиморфизмов у человека
Анализ и очистка чтений
Использовавшиеся команды
| Команда |
Комментарий |
| fastqc chr9_1.fastq |
Создает .html страницу с отчетом и .zip архив с содержимым страницы, а также логи. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 ../chr9_1.fastq 91-50.trimmed.fastq TRAILING:20 MINLEN:50 |
Убирает из рида нуклеотиды с phred score меньше 20 и удаляет риды, в которых удалилось больше половины
нуклеотидов(изначально длина всех ридов 100). |
Из предложенных двух .fastq файлов для девятой хромосомы был выбран chr9_1.fastq, так как в нем
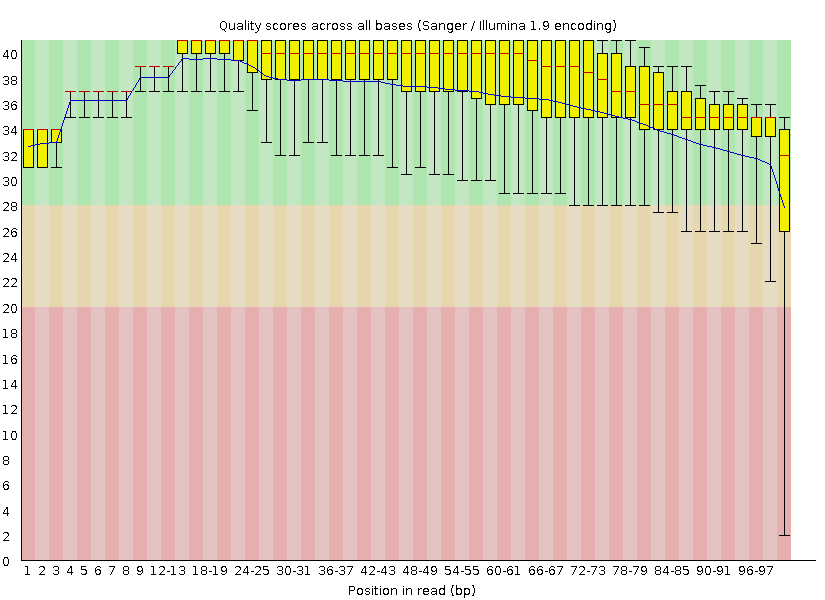
больше ридов. Ниже приведен Per base sequence quality из отчета fastqc для необработанного trimmomatic файла.
После очистки с помощью trimmomatic картина намного улучшается:
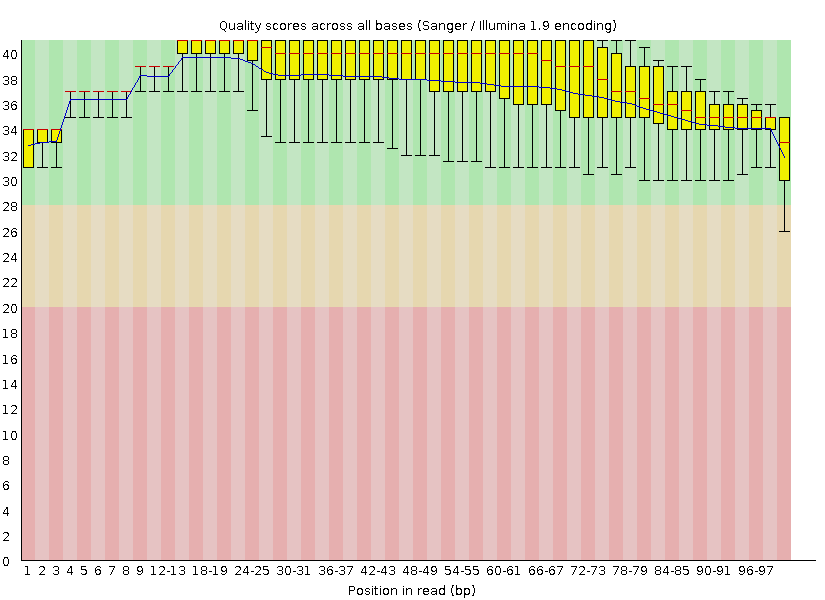
Стоит заметить, что число ридов после очистки сократилось всего на 165(было 10701, стало 10536): в число удаленных ридов попали риды, состоящие более чем наполовину(50 нуклеотидов) из оснований с
phred score меньше 20.
Картирование ридов
Использовавшиеся команды
| Команда |
Комментарий |
| PATH=$PATH:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Позволяет bash искать исполняемые файлы в указанной директории |
| hisat2-build chr9.fasta chr9 |
Индексирует референсную последовательность для последующей обработки |
| hisat2 -x chr9 -U ../trim/91-50.trimmed.fastq -S ../mapped_reads.sam --no-spliced-alignment --no-softclip 2> hisat2.log |
Накладывает риды на геном. -х — префикс .ht2 файлов, созданных hisat2-build, -U — файл со чтениями, -S выходной файл .sam,
--no-spliced-alignment указывает, что рид должен лечь непрерывно, --no-softclip не позволяет наложиться какой-то части рида, игнорируя то, что на концах
,"2>" перенаправляет вывод программы в лог-файл(hisat2 печатает лог на stderr). |
Анализ выравнивания
Использовавшиеся команды
| Команда |
Комментарий |
| samtools view -b -o mapped_reads.bam mapped_reads.sam |
Перевод .sam в бинарный вид .bam. |
| samtools sort mapped_reads.bam mapped_reads_sorted |
Сортирует выравнивания ридов в соответствии с их порядком в геноме |
| samtools index mapped_reads_sorted.bam |
Индексация выравниваний |
Вывод hisat2:
10536 reads; of these:
10536 (100.00%) were unpaired; of these:
73 (0.69%) aligned 0 times
10461 (99.29%) aligned exactly 1 time
2 (0.02%) aligned >1 times
99.31% overall alignment rate
Как видно отсюда, большинство(10461) ридов легли на геном ровно один раз. Не были наложены 73 рида и два наложились
более одного раза.
Поиск SNP и инделей
Использовавшиеся команды
| Команда |
Комментарий |
| samtools mpileup -uf chr9.fasta -o polymorph.bcf mapped_reads_sorted.bam |
Создает файл с полиморфизмами в формате .bcf, -u указывает на отсутствие сжатия выходного
файла, после -f идет референсная последовательность. |
| bcftools call -cv polymorph.bcf -o polymorph.vcf |
Делает из .bcf .vcf. |
В таблице ниже описаны три полиморфизма из файла .vcf.
| # |
Координата |
Тип полиморфизма |
Референс/Чтение |
Глубина покрытия |
Качество чтений |
| 1 |
4985879 |
SNP |
A/G |
30 |
225.009 |
| 2 |
5090641 |
SNP |
G/A |
98 |
221.999 |
| 3 |
136133380 |
INDEL |
CGGGG/CG |
-* |
214.475 |
*Не указано.
Итого получилось 111 полиморфизмов: 5 инделей и 106 SNP.
С помощью MS excel получили информацию о покрытии и качестве прочтений SNP. Покрытие варьирует от 1 рида до 98, среднее покрытие — 12,16981132(медиана 3), качество варьирует
от 3,54557 до 226,13, среднее значение — 79,604775(медиана 28,39195). Таким образом, риды распределяются по геному неравномерно.
Аннотация SNP
Базы данны для аннотации были найдены по адресу /nfs/srv/databases/annovar/humandb.old/.
Использовавшиеся команды
| Команда |
Комментарий |
| vcftools --vcf polymorph.vcf --remove-indels --recode --recode-INFO-all --out poly_SNP |
Избавляемся от инделей. --vcf — название входного файла, --remove-indels — то что мы собственно просим,
--recode и --recode-INFO-all позволяет сохранить поле INFO в полученном vcf файле(по дефолту оно удаляется). |
| convert2annovar.pl -format vcf4 poly_SNP.vcf > poly_SNP.avinput |
Переводит .vcf в формат annovar. |
| annotate_variation.pl -filter -out snp138_filtered -build hg19 -dbtype snp138 poly_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по dbSNP. |
| annotate_variation.pl -out refGene_filtered -build hg19 poly_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по RefGene. |
| annotate_variation.pl -filter -out 1000g -build hg19 -dbtype 1000g2014oct_all poly_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по 1000 Genomes. |
| annotate_variation.pl -regionanno -out gwas -build hg19 -dbtype gwasCatalog poly_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по GWAS. |
| annotate_variation.pl -filter -out clinvar -build hg19 -dbtype clinvar_20150629 poly_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по ClinVar. |
RefSeq делит snp по их расположению в гене на следующие категории:
| Категория |
Комментарий |
| exonic |
Вариант в кодирующем экзоне |
| splicing |
Вариант в 2 парах оснований от сайта сплайсинга |
| ncRNA |
Вариант в транскрипте, для которого в данной аннотации (refSeq, Ensembl, etc.)
не указано наличие кодирующего региона |
| UTR5 |
Вариант в 5'-нетранслируемой области |
| UTR3 |
Вариант в 3'-нетранслируемой области |
| intronic |
Вариант в интроне |
| upstream |
Вариант на расстоянии 1kb от старта транскриптиции(в сторону 5' конца) |
| downstream |
Вариант на расстоянии 1kb от конца транскриптиции(в сторону 3' конца) |
| intergenic |
Вариант в межгенной области |
Число наших SNP в найденных категориях(файл variant_function):
| Категория |
Число |
| UTR3 |
8 |
| exonic |
15 |
| intronic |
78 |
| downstream |
2 |
| intergenic |
3 |
Также refGene дает информацию о (не)синонимичности SNP(файл exonic_variant_function), правда из 15 экзонных мутаций были данные только по трем:
| SNP |
Комментарий |
| chr9 5081780 5081780 G A hom 221.999 50 |
synonymous SNV JAK2:NM_004972:exon19:c.G2490A:p.L830L |
| chr9 5126443 5126443 T A het 207.009 38 |
nonsynonymous SNV JAK2:NM_004972:exon24:c.T3288A:p.D1096E |
| chr9 6253571 6253571 C T het 225.009 54 |
synonymous SNV IL33:NM_033439:exon6:c.C489T:p.Y163Y,IL33:NM_001199641:exon3:c.C111T:p.Y37Y,IL33:NM_001199640:exon5:c.C363T:p.Y121Y* |
*Для разных транскриптов возможны разные координаты.
SNP принадлежат пяти генам: ABO, GLIS3, IL33, JAK2, SLC1A1.
96 из 106 имеют rs(перечислены в выводе при аннотации dbSNP).
Из аннотации 1000 Genomes можно получить данные по частотам SNP. Cредняя частота составила 0.41. Причем есть один очень странный
SNP с частотой 1: 1 chr9 6253186 6253186 C G hom 145.033 8
3 аннотированы в ClinVar. Но больше ничего про них не указано в выводе Annovar. Зато в GWAS есть информация
о клинической картине.
| Предрасположенность/фенотипический признак | SNP |
|---|
| Crohn's disease |
chr9 4985879 4985879 A G het 225.009 30 |
| Endometriosis | chr9 6253571 6253571 C T het 225.009 54 |
| Tumor biomarkers,Coagulation factor levels | chr9 136131188 136131188 C T het 13.2188 10 |
| mean corpuscular hemoglobin concentration | chr9 136131322 136131322 G T het 180.009 24 |
| End-stage coagulation | chr9 136131415 136131415 C T het 134.008 21 |
| Malaria | chr9 136132754 136132754 C A het 123.008 33 |
| Activated partial thromboplastin time,Venous thromboembolism,D-dimer levels | chr9 136137065 136137065 A G hom 42.7648 2 |
| Coagulation factor levels | chr9 136137106 136137106 G A hom 9.52546 1 |
Полную таблицу по всем SNP можно скачать по ссылке.
|