BLAST
Что было сделано:
- 1. были определены таксономия и функция прочтённой нуклеотидной последовательности (из практикума 6)
- 2. было проведено сравнение списков находок нуклеотидных последовательностей тремя разными вариантами blast - megablast, blastn (default), blastn ('sensitive')
- 3. было проверено наличие гомологов трех белков в неаннотированном геноме
- 4. был найден ген белка в одном из контигов
Задание 1
Чтобы определить таксономию и функцию нуклеотидной последовыательности, полученной путем секвенирования по Сэнгеру, нужно найти наиболее близкого гомолога. Сделать это, по предположению, получится с помощью алгоритма megablast.
Параметры поиска брали по умолчанию.
Алгоритм показал 100 последовательностей.
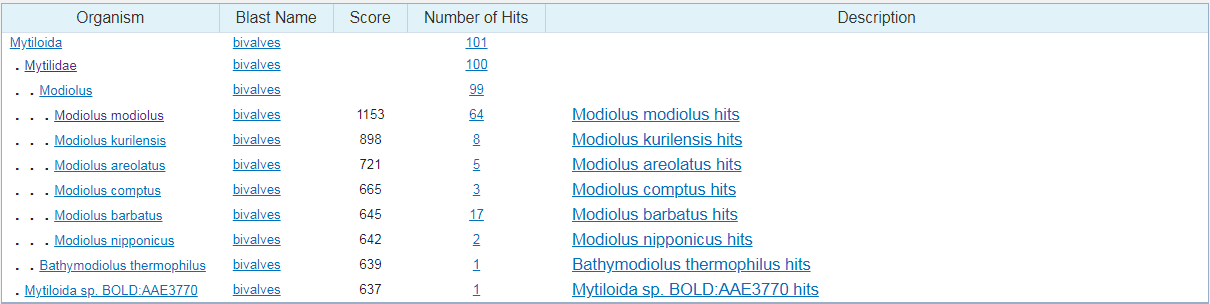
Почти все результаты выдали ген первой субъединицы цитохром с-оксидазы, обозначаемой у организмов эукариот как СОХ1, СО1 или СОI. Были несколько результатов с полным митохондриальным геномом.
Все выданные записи принадлежат организмам семейства митилид - морским двустворчатым моллюскам, распространенным по всему Мировому океану.
Наиболее подходящим организмом, судя по Score (чем больше, тем лучше), E-value ~0.0, проценту идентичности (больше 99.0%), проценту покрытия (больше 90%), наиболее подходящим организмом является Modiolus modiolus vouscher.
Таким образом, с помощью алгоритма megablast можно выявить функцию последовательности и таксономию хозяина этой последовательности. В нашем случае последователльность кодирует цитохром с-оксидазу - оксидаза аэробной дыхательной цепи переноса электронов. Предполагаемая таксономия организма:
- Надцарство Eukariota
- Царство Metazoa
- Тип Mollusca
- Класс Bivalvia
- Подкласс Pteriomorphia
- Порядок Mytiloida
- Надсемейство Mytiloidea
- Семейство Mytilidae
- Подсемейство Modiolinae
- Род Modiolus
- Вид Modiolus modiolus
Примечание: Картинки с результатами работы с бластом приведены ниже:
Предполагаемые организмы:

Покрытие:

Предполагаемая таксономия:
Задание 2
Чтобы сравнить результаты выдачи трех вариантов blast, предлагается сравнить сначала по своей последовательности из предыдущего задания. Предложено было 3 варианта работы алгоритма: megablast, blastn (параметры по умолчанию), blastn ("чувствительные" параметры)
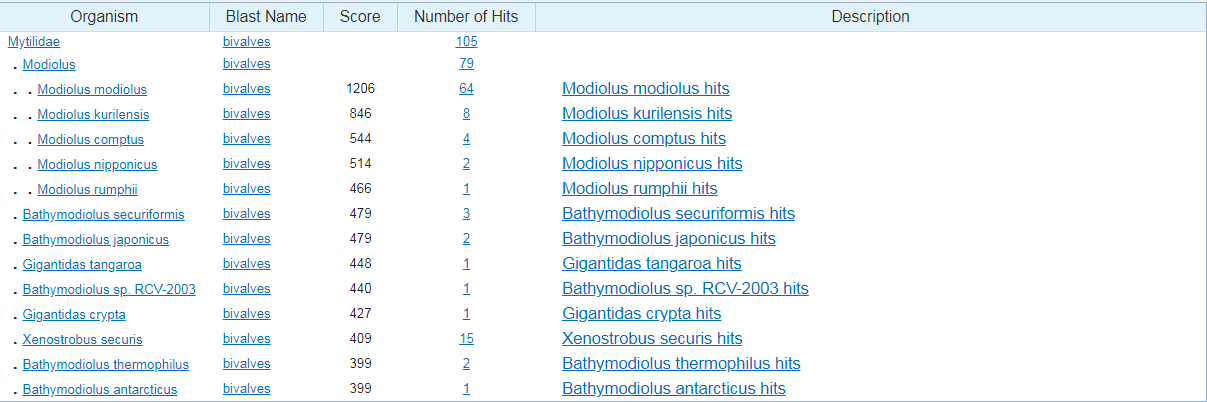
Начнем с первого - megablast. Просто вбиваем консенсусную последовательность и бластуем с параметрами по умолчанию. Получилось то же, что и в задании 1, разумеется, ведь использован был тот же самый алгоритм и та же самая последовательность. Число находок 100. Картинки можно посмотреть выше.
Теперь blastn с параметрами по умолчанию. Делаем то же самое, что и в предыдущем опыте, только изменяем алгоритм. Число находок, по-прежнему, 100. Длины последовательностей в выравнивании даже более схожи, чем при использовании megablast.
Предполагаемые организмы:

Покрытие:

Предполагаемая таксономия:
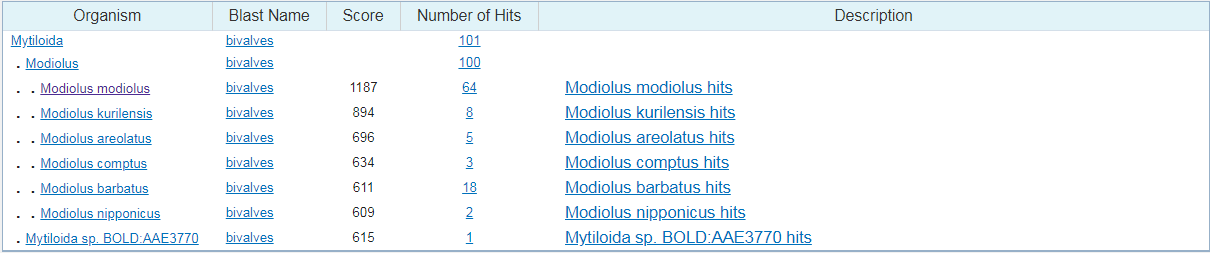
Дальше чуть поинтереснее, наверное: blastn с чувствительными параметрами. Чтобы сделать параметры алгоритма более чувствительными, уменьшим длину слова до 7 и выберем значение Match/Mismatch Scores на 1/-4. Все то же количество находок - 100, однако встретились последовательности, процент покрытия которых не внушает доверие. Думаю, были подобраны не самые чувствительные параметры.
Предполагаемые организмы:

Покрытие:

Предполагаемая таксономия:
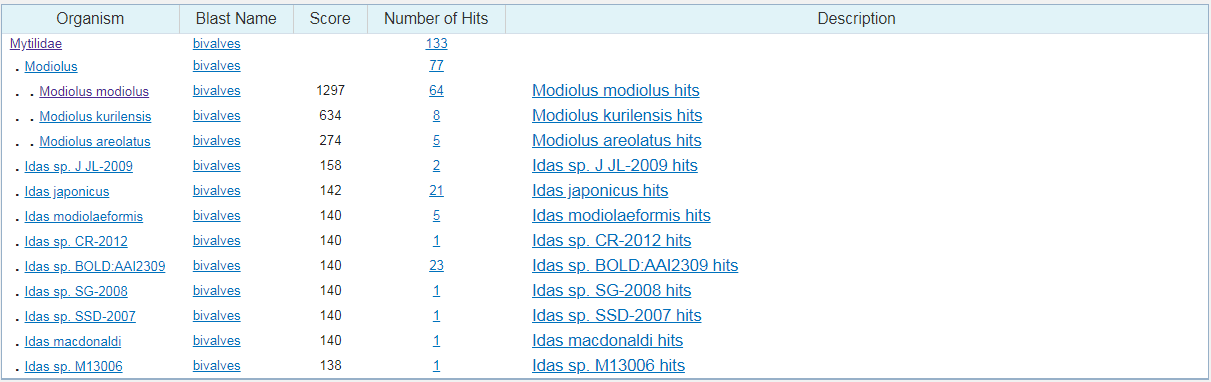
Попробую сменить параметр Match/Mismatch Scores на 4/-5, остальное оставить неизменным. Результаты в разы лучше, и теперь они больше похожи на результаты работы предыдущих алгоритмов. Процент покрытия последовательностей в выравнивании стал чуть больше, число находок, по-прежнему, 100.
Предполагаемые организмы:
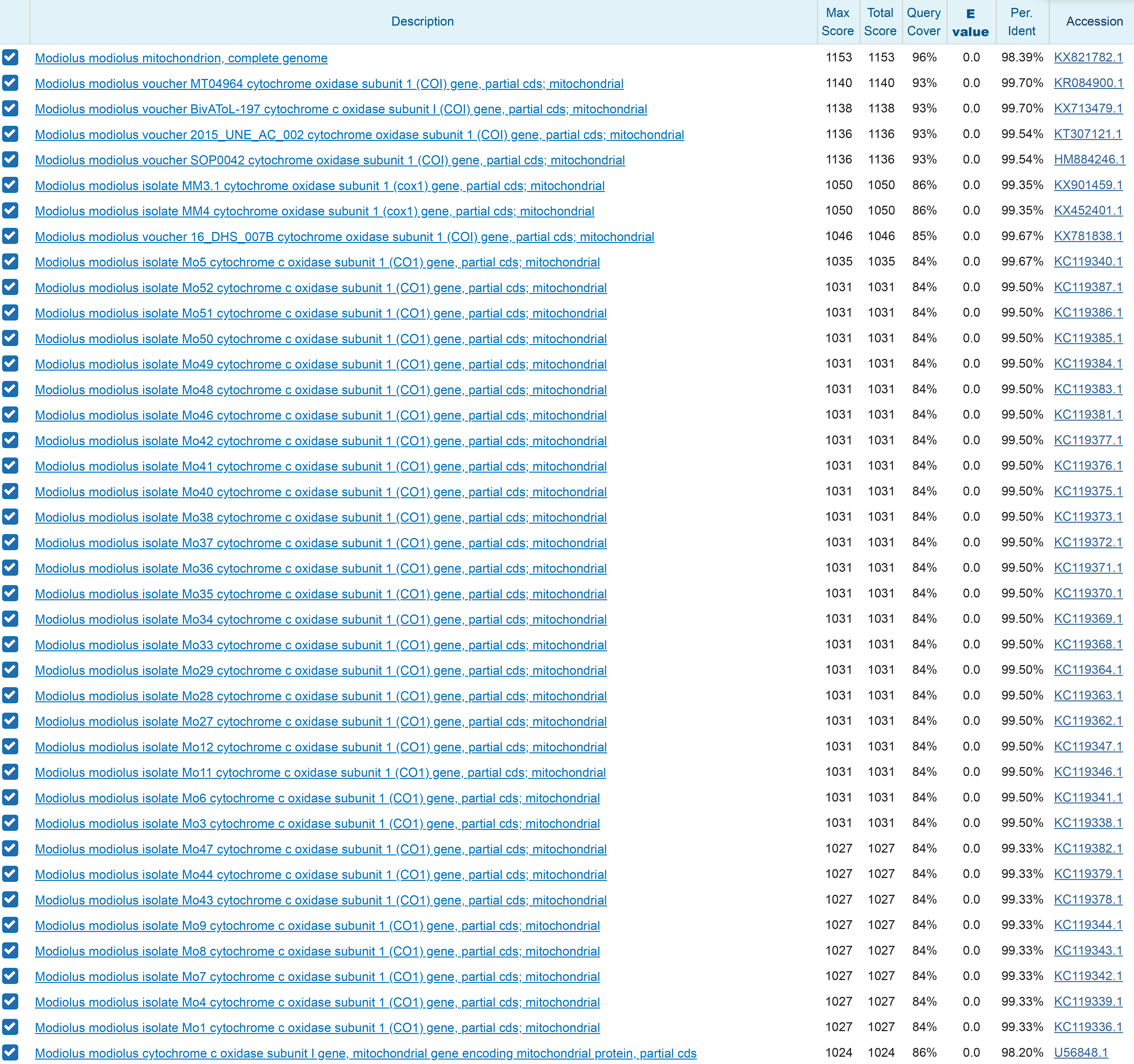
Покрытие:

Предполагаемая таксономия:
Примечаниe: Скачать консенсусную последовательность из шестого практикума можно здесь
Далее предлагается проделать ту же работу, только с одним из CDS из 7 практикума. Я взяла самый первый CDS из этого файла.
megablast (параметры по умолчанию): алгоритм выдал 13 находок - среди них есть фаг из 7 практикума, остальные последовательности принадлежат, в основном, Спироплазмам. Процент покрытия и идентичности у болльшинства последовательностей достаточно большой - около 90%, но у некоторых довольно короткое покрытие (отмечено зеленым на картинке с покрытиями.)
Предполагаемые организмы:
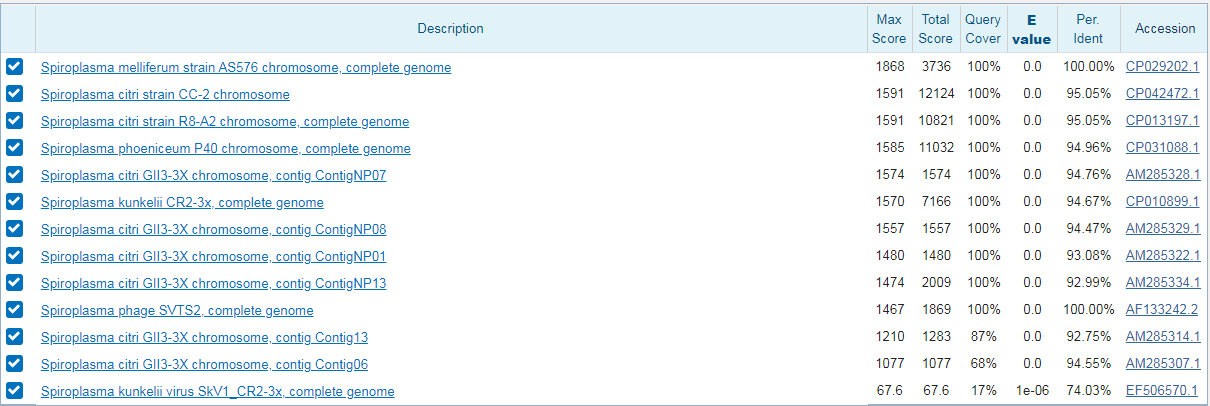
Покрытие:
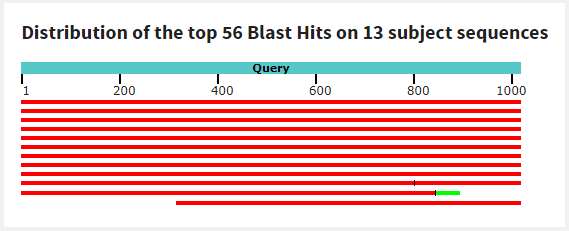
Предполагаемая таксономия:
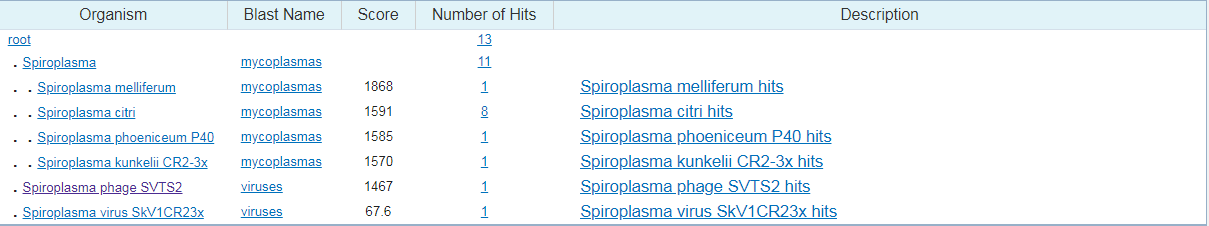
blastn с параметрами по умолчанию: алгоритм выдал 100 находок - среди них есть те 2 последовательности, которые принадлежат разным организмам (Спироплазме и ее фагу), однако имеют 100% покрытие и идентичность (эти последовательности были выданы и при пользовании алгоритмом megablast). Большинство из представленных последовательностей с неприлично маленьким покрытием (менее 10%) и большим E-value. Если не брать такие последовательности в расчет, то остается всего 12 последовательностей, которые нам удовлетворяют. Почти у всех из удовлетворяющих последовательностей покрытие 100%, E-value~0, а идентичность около 100%, что очень даже неплохо. Как и в поиске мегабластом, хозяевами последовательностей в большинстве случаев является Спироплазма.
Предполагаемые организмы:
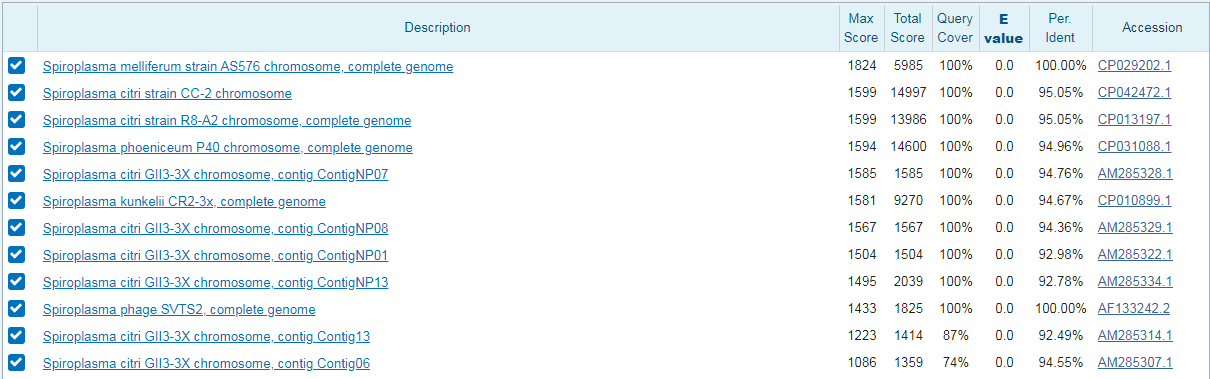
Покрытие:
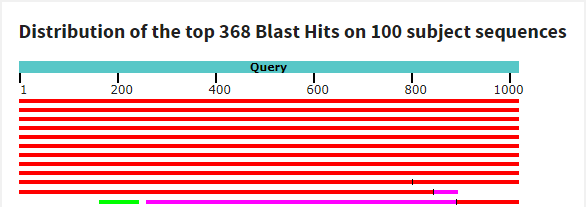
Предполагаемая таксономия:
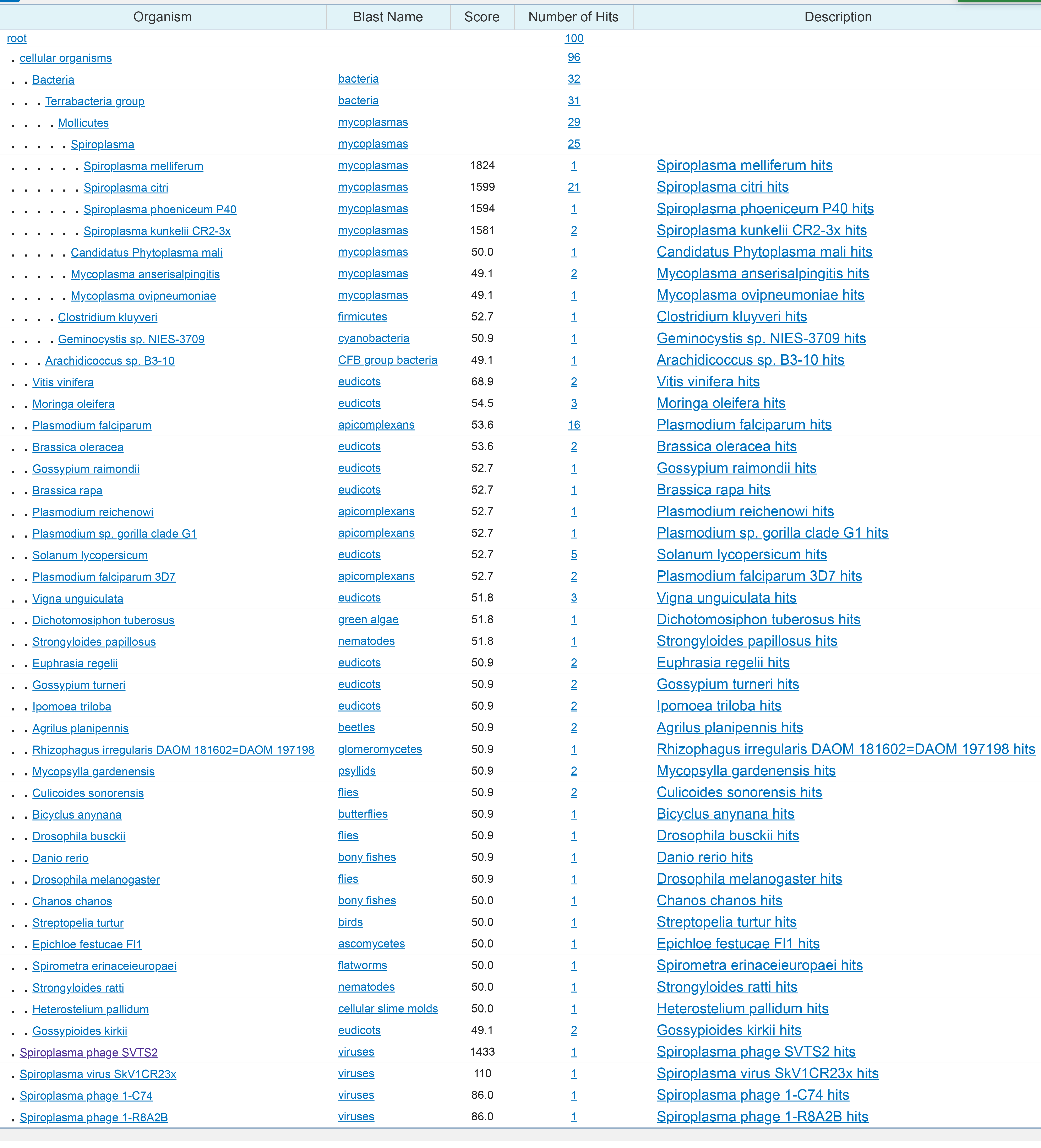
blastn c Match/Mismatch Scores 4/-5 и длиной слова 7: выдано 100 находок, они сравнительно лучше выданных предыдущим алгоритмом, по-прежнему удовлетворяют поиску по покрытию, идентичности и E-value почти те же 12 последовательностей.
Предполагаемые организмы:
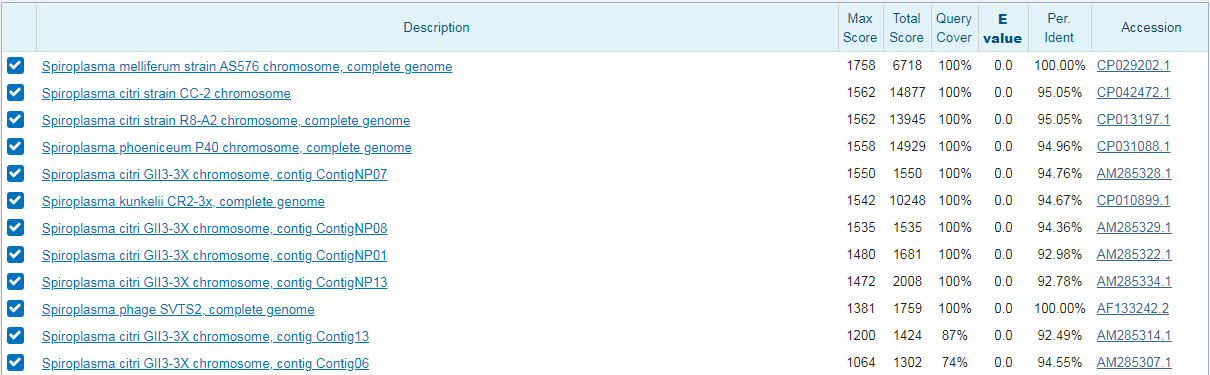
Покрытие:
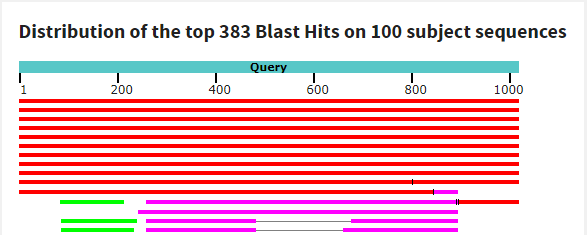
Предполагаемая таксономия:
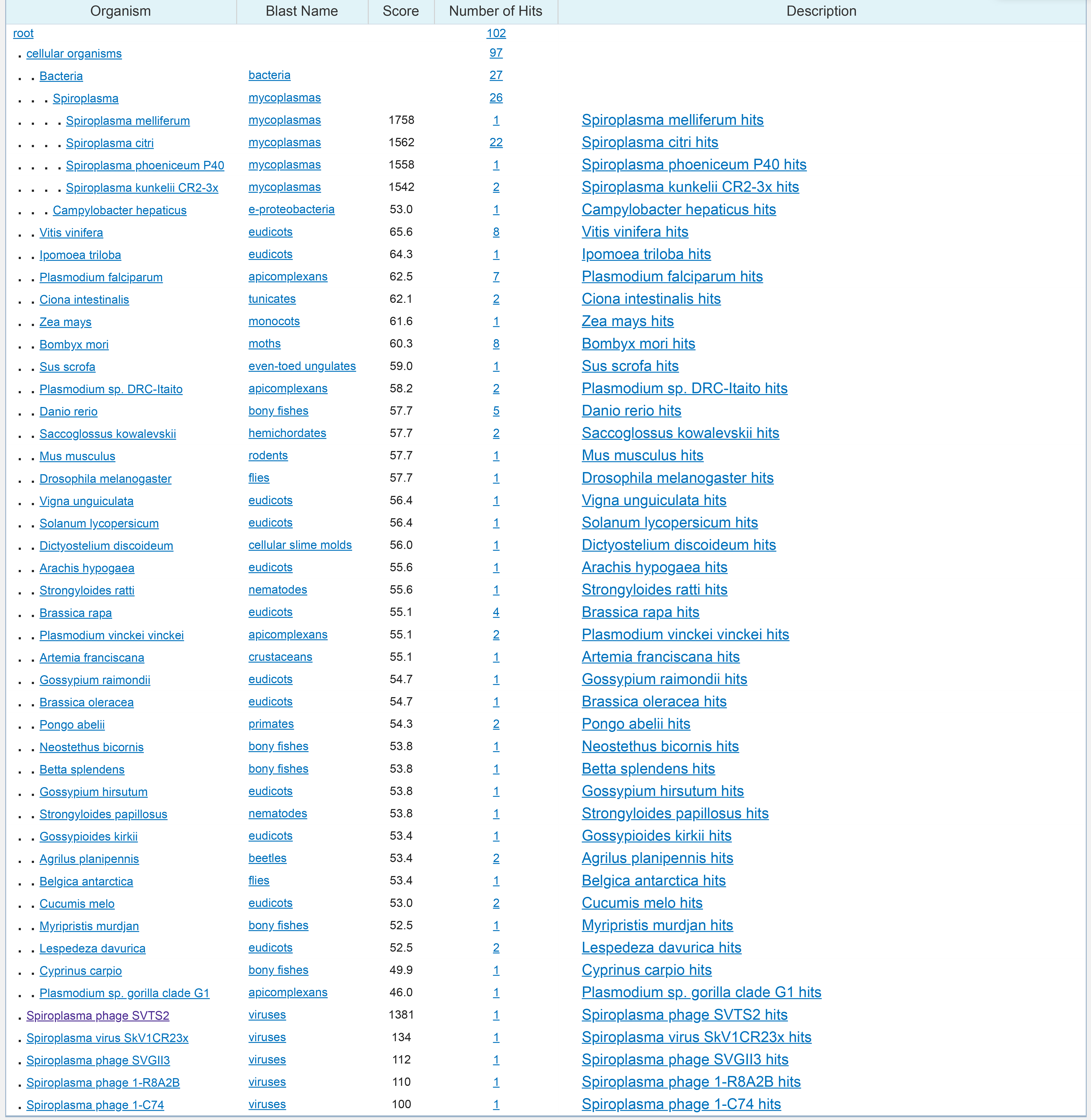
В целом, megablast ищет наиболее близких гомологов и делает это достаточно быстро, а blastn находит больше последовательностей, но они не все могут называться гомологичными. Также были опытным путем установлены наиболее чувствительные параметры blastn: Match/Mismatch Scores 4/-5 и длина слова 7.
Задание 3
Требовалось проверить наличие гомологов трех белков в неаннотированном геноме Amoeboaphelidium protococcarum. Чтобы найти гомологи белков в формальной трансляции, нужно использовать алгоритм tblastn. Я выбрала три белка, которые, по предположению, должны быть распространены у всех эукариот:
- RPB2 - фермент, катализирующий транскрибцию ДНК и синтезирующий предшественников мРНК
- elF2 - фактор инициации трансляции у дрожжей, участвующий в выборе инициирующего кодона
- DNA ligase [Thermus thermophilus HB8] - фермент, катализирующий образование фосфодиэфирной связи в однонитевом разрыве ДНК
Через терминал на кодомо были созданы три файла с результатами работы tblastn
elF2
tblastn -query elf2.fasta -db genome.fasta -out elf2.out -evalue 0.01 -outfmt 7
# TBLASTN 2.2.28+ # Query: XP_001383055.2 eIF2 [Scheffersomyces stipitis CBS 6054] # Database: genome.fasta # Fields: query id, subject id, % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score # 2 hits found XP_001383055.2 scaffold-51 50.31 159 75 2 111 269 20325 20789 3e-41 154 XP_001383055.2 scaffold-104 50.31 159 75 2 111 269 357434 357898 9e-41 152 # BLAST processed 1 queries
Эти находки, скорее всего, можно назвать гомологами, так как процент идентичности >=50, покрытие хорошее за счет малого числа гэпов, E-value достаточно мало, чтобы рассматривать эти последовательности как гомологи искомой последовательности белка.
DNA ligase [Thermus thermophilus HB8]
tblastn -query dna_ligase.fasta -db genome.fasta -out dna_ligase.out -evalue 0.01 -outfmt 7
# TBLASTN 2.2.28+ # Query: AAA27486.1 DNA ligase [Thermus thermophilus HB8] # Database: genome.fasta # Fields: query id, subject id, % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score # 1 hits found AAA27486.1 unplaced-982 46.91 81 40 3 569 646 18475 18717 8e-09 58.9 # BLAST processed 1 queries
Эту находку, я думаю, нельзя назвать гомологичной последовательности искомой ДНК-лигазы, потому что длина выравнивания ну уж очень мала по сравнению с последовательностью искомого белка.
RPB2
tblastn -query rpb2.fasta -db genome.fasta -out rpb2.out -evalue 0.01 -outfmt 7
# BLASTN 2.2.28+ # Query: AFV31136.1 RPB2 [Amoeboaphelidium protococcarum] # Database: /home/students/y18/spyro/term3/block2/pr8/genome.fasta # 0 hits found # BLAST processed 1 queries
Гомолог не нашелся, что очень странно. Именно эта последовательность должна быть в геноме Amoeboaphelidium protococcarum. Вот ссылка в NCBI. Возможно, это связано с тем, что база данных на kodomo слегка устаревшая.
Задание 4
Возьмем какой-нибудь скэффолд, скажем, 0, взятый из файла Х5 из предыдущего задания. Используем blastx, который выравнивает транслированную последовательность нуклеотидов с базой данных белков. Будем искать в базе данных RefSeq.
Длина искомой последовательности 2822.
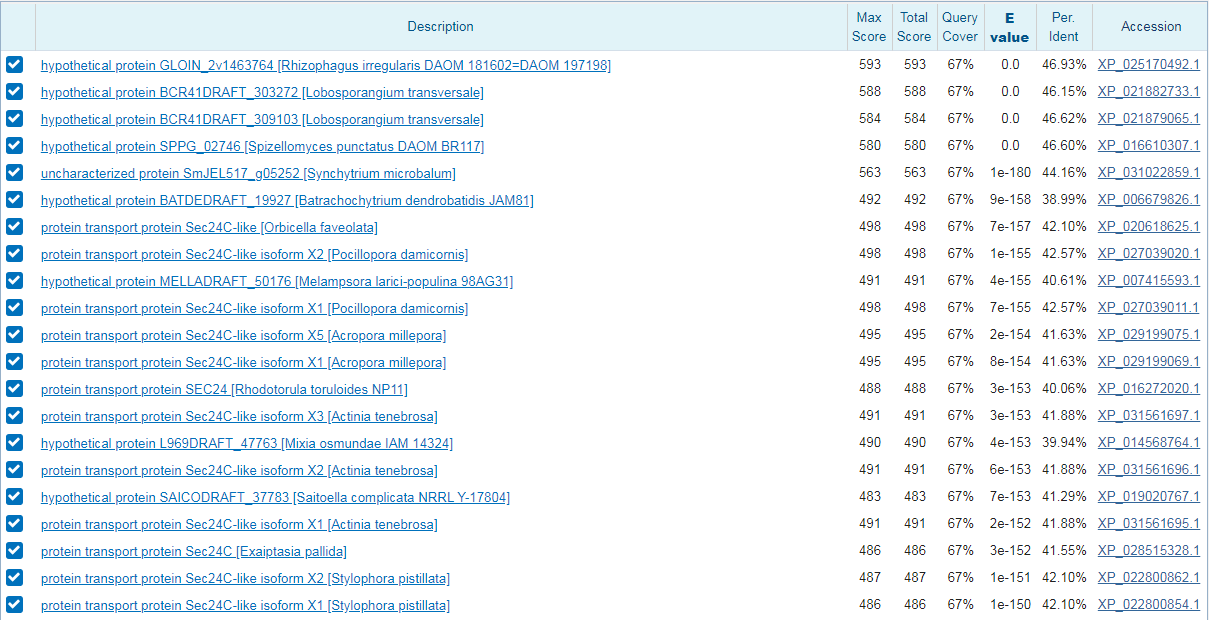
Алгоритм выдал 100 последовательностей с одним и тем же процентом покрытия (67%), но с немного разным E-value, у некоторых последовательностей оно настолько мало, что отображается просто как 0.0. Абсолютное большинство результатов выдало белок, участвующий в белковом транспорте. Имя этому белку Sec24C.
Результат работы алгоритма blastx можно увидеть на картинке ниже.