Исследованная бактерия - Raoultella ornithinolytica (CP010557.1)
Сайты считались избегаемыми при значениях отношения Карлина (контраста) меньше 0,78
Выходной файл веб-сервиса для генома бактерии
Избегаемые сайты РМ - там всего два сайта: CTAG, CCTAGG
Выходной файл веб-сервиса для набора контигов штамма из метагенома
Избегаемые сайты РМ согласно набору контигов штамма из метагенома - там всего один сайт: CTAG
Сайт CTAG избегается и согласно анализу полного генома, и набора контигов. В полном геноме сайтов найдено больше: найден еще один сайт CCTAGG. Это можно объяснить тем, что бактерия в кишечнике подвергается значительно меньшему воздействию фагов и потому ей, возможно, потеряла один из сайтов РМ в процессе эволюции.
Исследованная бактерия - Dyadobacter fermentans dsm 18053. В геноме бактерии 5717 генов. В результате была составлена выборка из 500 генов с длиной CDS больше 300 и аннотацией, отличной от 'hypothetical protein' (чтобы добиться нахождения достоверных мотивов). Для каждого гена с помощью скрипта был вырезан участок от -16 до -1 позиции до начала кодирующей последовательности.
С помощью программы MEME был осуществлен поиск мотивов de novo в вырезанных фрагментах. Установленные параметры: длина мотива от 4 до 6, 0 или 1 мотив на последовательность, искать мотив на той же цепи. Был произведен поиск 2 наилучших мотивов для сравнения e-value. Результат представлен на рисунке 1.
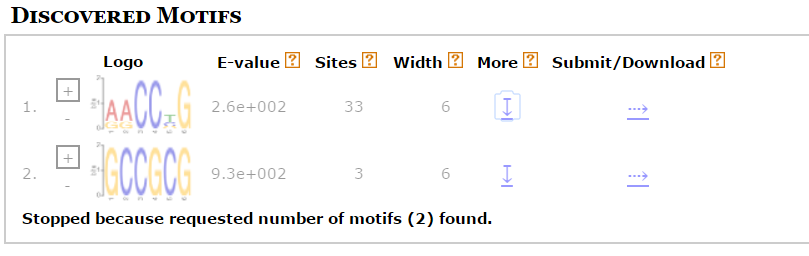
Рис. 1. Два мотива, найденных программой MEME в промоторных областях генов бактерии.
Нечего и говорить, что полученные E-value удручают. Изменение параметров (увеличение возможной длины мотива, поиск мотивов в случайной выборке из генов, поиск по всем генам и многие другие) к лучшему результату не привели. В результате поиска информации в литературе была найдена, например, следующая статья, в которой утверждается, что бактерии клады Bacteroidetes (представителем которой является D. fermentans), видимо, лишены SD.
Затем я произвел поиск в MEME с теми же параметрами, но уже только одного наилучшего мотива. Также я сохранил лого найденного MEME мотива (рисунок 2).
Рис. 2. Лого найденной последовательности Шайна-Дальгарно.
С помощью программы FIMO я произвел поиск найденного мотива для всех остальных генов бактерии. Для этого были вырезаны участки от -16 до -1 позиции до начала кодирующей последовательности у всех генов. Длина участков была увеличена, чтобы немного снизить вероятность ошибок поиска для неправильно аннотированных генов. В результате для 5717 генов было найдено 6042 мотива (с p-value меньше 0.1). Такое большое число находок может быть связано с наличием случайных последовательностей, так как сигнал SD достаточно слабый. Среди найденных 910 последовательностей имело p<0.01.
Затем по результатам работы FIMO была построена гистограмма распределения начала найденных SD от старта трансляции. Полученное изображение представлено на рисунке 3.
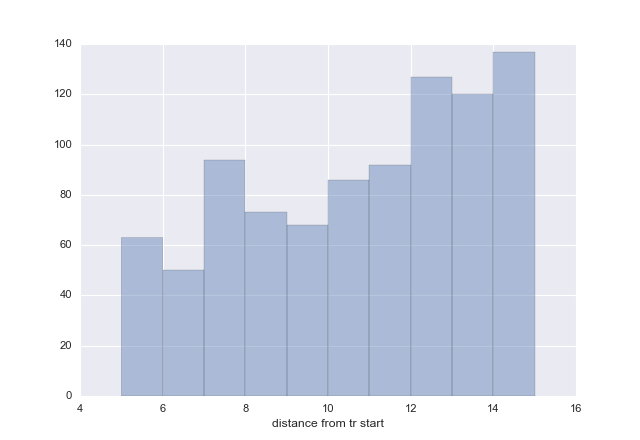
Гистограмма распределения начала найденных SD от старта трансляции для порого p-value 0.01 (16% генов). Числа на оси X означают расстояние от начала SD до старта трансляции.
Видимо, у данной бактерии действительно нет SD, и, как предполагают авторы статьи выбор стартов трансляции происходит при связывании рибосомного белка S1 с неструктурированной 5' UTR (GC-контент у Bacteroidetes заметно ниже обычно наблюдаемого в промоторной области).
Качество данных было проверено с помощью FastQC. Вот : результат Всего ридов 26325. Как видно из рисунка, качество чтения достаточно высокое на всём протяжении последовательности. Поэтому программа Trimmomatic для очистки чтений не применялась.
Затем чтения были откартированы на геном человека hg19 (заранее проиндексированный) с помощью команды bwa mem ../../hg19/GRCh37.p13.genome.fa chipseq_chunk42.fastq > chipseq_chunk42.sam. Далее были использованы следующие команды: samtools view -bSo chipseq_chunk42.bam chipseq_chunk42.sam (переводит выравнивание чтений с референсным геномов в бинарный формат, с которым потом работают программы), samtools sort chipseq_chunk42.bam -T chip_temp -o chipseq_chunk42.sorted.bam (сортирует выравнивание по координате начала чтения в референсе), samtools index chipseq_chunk42.sorted.bam (индексирует отсортированный файл), samtools idxstats chipseq_chunk42.sorted.bam > chipseq_chunk42.idxstats (записывает в файл chipseq_chunk42.idxstats информацию о количестве чтений, откартированных на каждый элемент генома) и samtools view -c chipseq_chunk42.sorted.bam (показывает, сколько чтений в сумме было откартированно на все элементы генома).
Всего было 26325 чтений, все они были откартированы на геном. При этом распределение чтений по хромосомам позволяет предположить, что я работаю с участками 20 хромосомы человека.
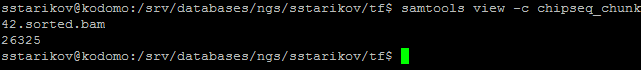
Рис. 5. Число чтений, откартированных на геном, по результатам работы описанных программ.
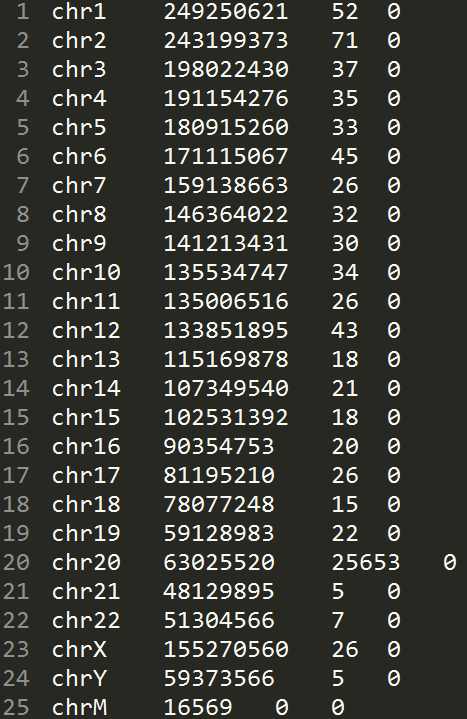
Рис. 6. Распределение числа откартированных чтений по хромосомам.
Далее я произвел поиск пиков с помощью программы MACS. Так как пиков было найдено 0, использовалась команда macs2 callpeak -t chipseq_chunk42.sorted.bam -n chipseq_chunk42 --nomodel. Были получены следующие файлы: chipseq_chunk42_peaks.narrowPeak , chipseq_chunk42_peaks.xls и chipseq_chunk42_summits.bed.
Всего найдено 13 пиков. Общее их расположение представлено на рисунке 7, а 3 пика в отдельности - на рисунках 8-10. Длина пика указана в подписи к соответствующему рисунку.
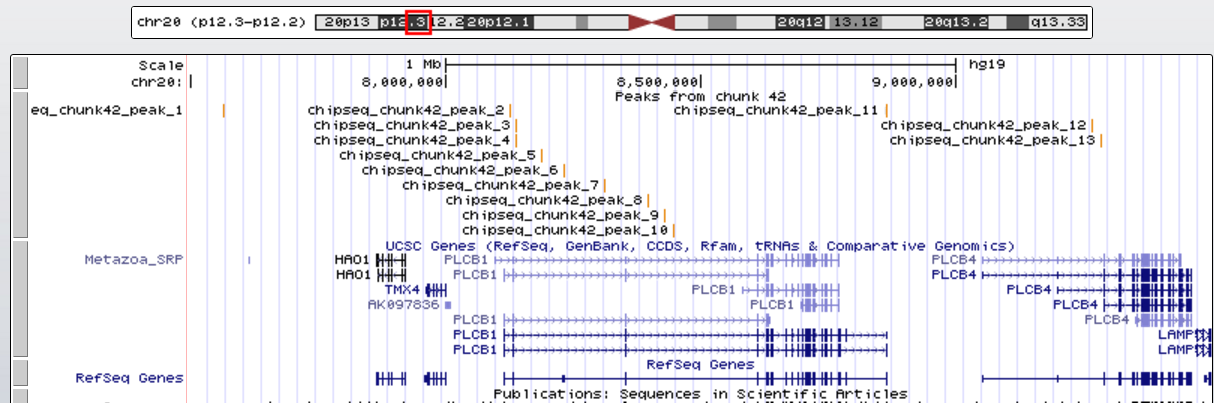
Рис. 7. Расположение найденных пиков в геноме человека (сборка hg19) (UCSC)
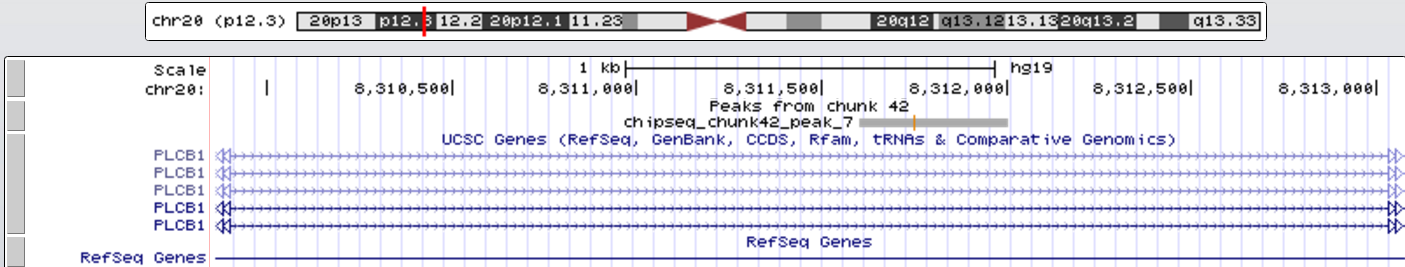
Рис. 8. Увеличенный 7 пик, занимающий позицию chr20:8311603-8312006 и имеющий длину 404 нт.
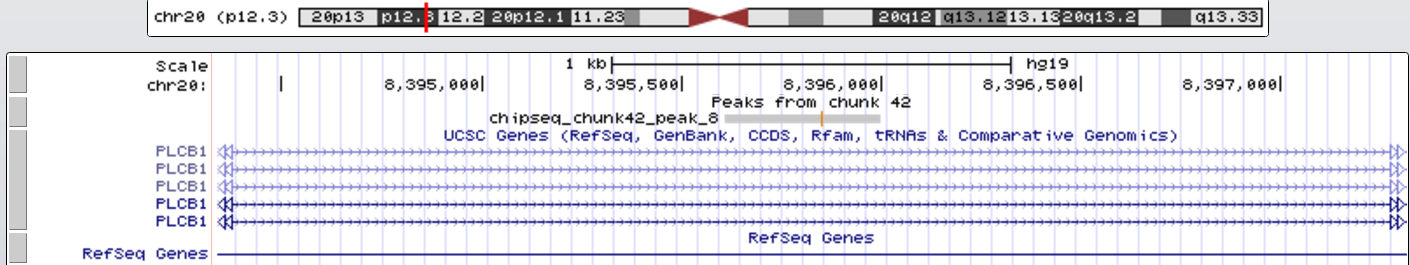
Рис. 9. Увеличенный 8 пик, занимающий позицию chr20:8395609-8395998 и имеющий длину 390 нт.
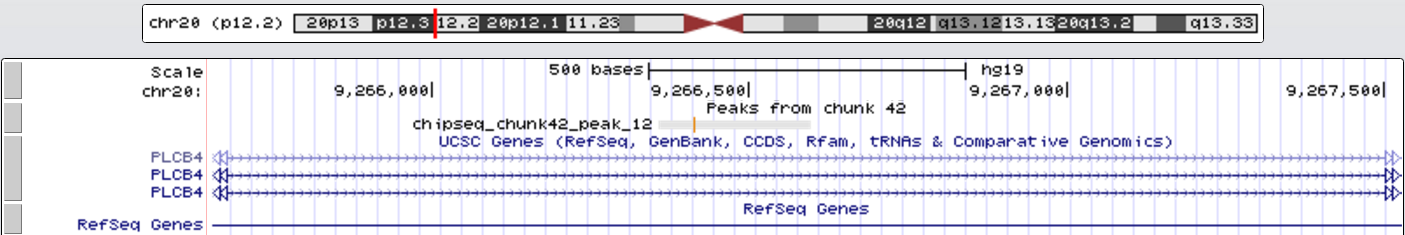
Рис. 10. Увеличенный 12 пик, занимающий позицию chr20:9266359-9266596 и имеющий длину 238 нт.
Все пики имеют достаточно высокий -log10(p_value) (и -log10(q_value) тоже), а значит являются достоверными. Пики 7, 8 попали внутрь гена PLCB1, а пик 12 - внутрь гена PLCB4. У пиков 7, 12 вершина сдвинута влево относительно середины, а у пика 8 - вправо.
TBP является одним из ключевых ДНК-узнающих белков, необходимых для образования на промоторах генов комплекса TFIID и инициации транскрипции с помощью Pol II. Тем не менее, лишь часть промоторов имеет сигнал TATA-box, связываемый TBP. Консенсусная последовательность для связывания TBP - TATAWAAR.
Выбранный эксперимент - ChIP-seq анализ для TBP в стволовых эмбриональных клетках человека H1-hESC с использованием антител кролика.
Было описано три гена, транскрипция которых инициируется с помощью TBP, и один - без сигнала TATA-box в промоторной области.
Первый ген с TATA-box - CDC40 (участвует в регуляции клеточного цикла, фактор процессинга пре-мРНК). Его координаты chr6:110501624-110553422, длина 51799, + цепь, 15 экзонов. TATA-box на расстоянии около 60 нт.
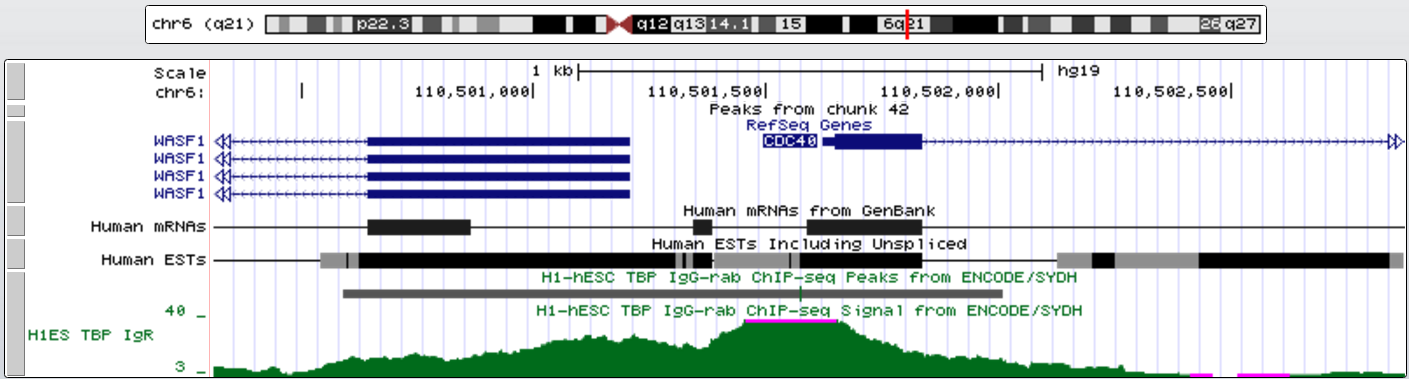
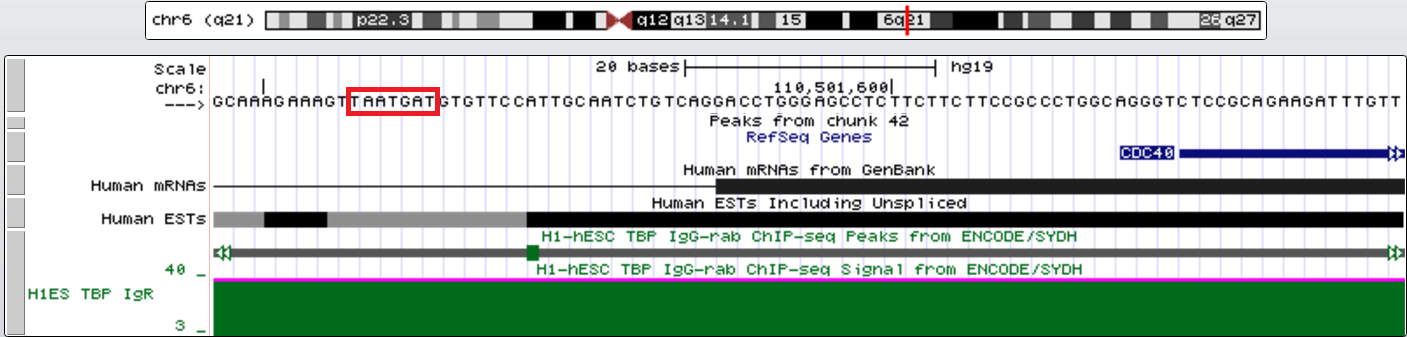
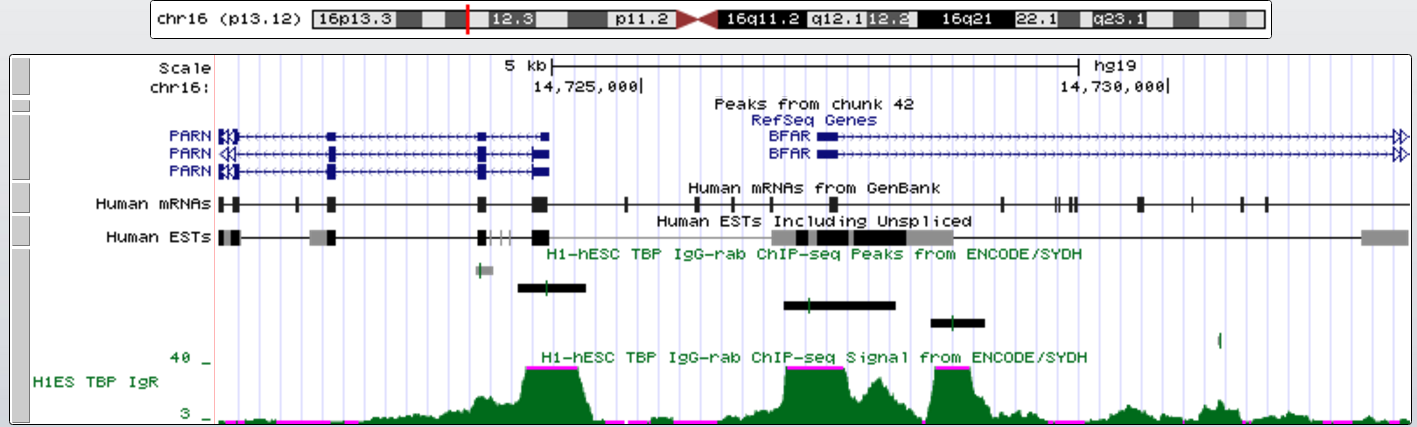
Второй ген с TATA-box - PARN (polyA-специфичная рибонуклеаза). Его координаты chr16:14529557-14724128, длина 194572, - цепь, 24 экзона. TATA-box на расстоянии около 60 нт.
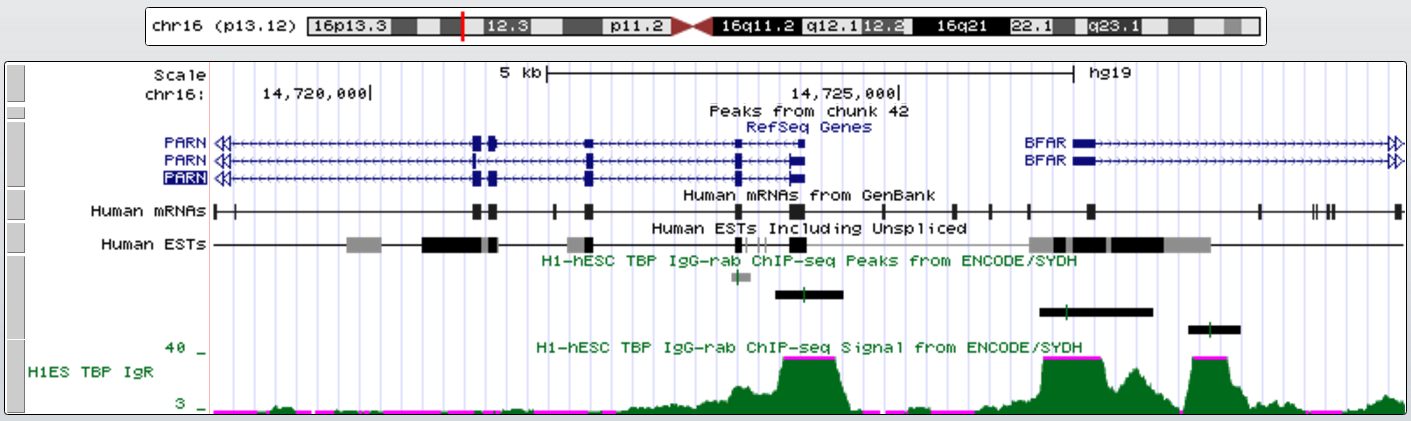
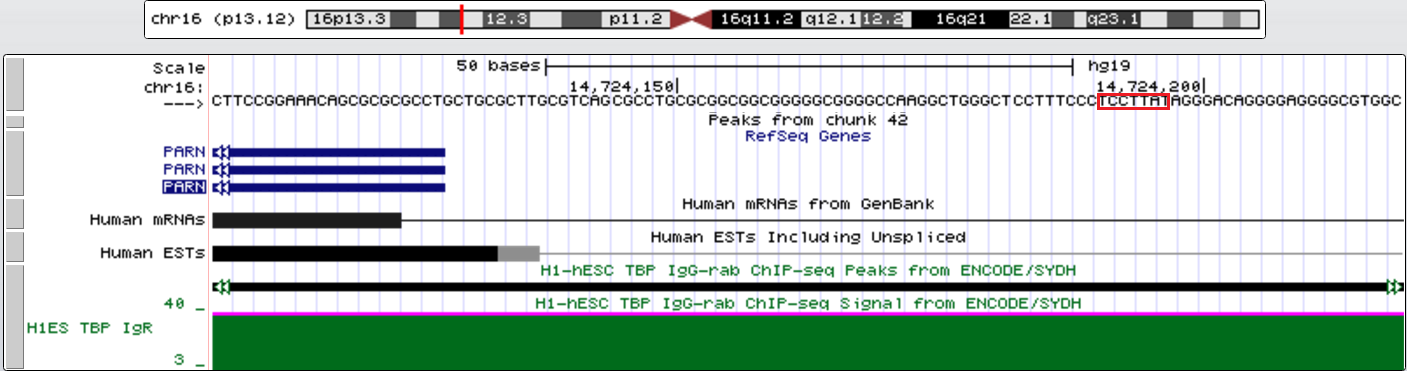
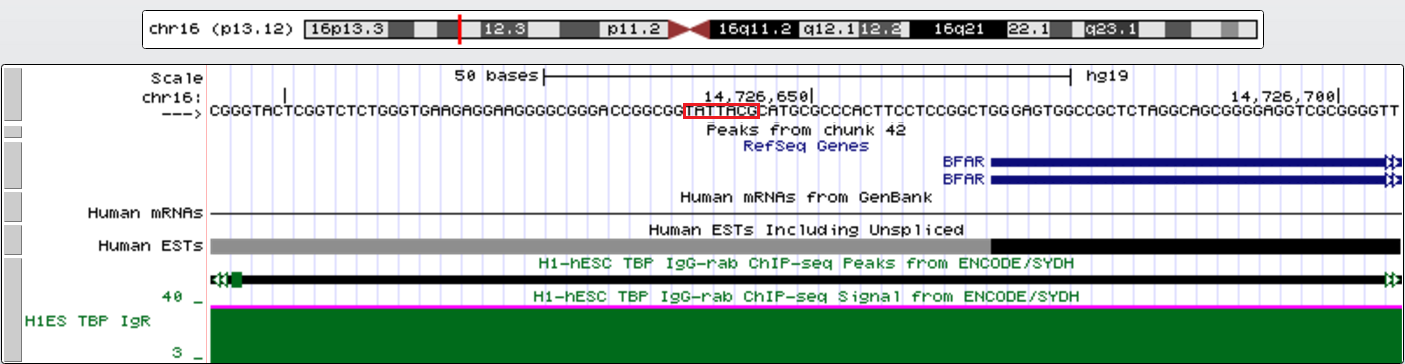
Третий ген с TATA-box - BFAR (бифункциональный регулятор апоптоза). Его координаты chr16:14726668-14763093, длина 36426, + цепь, 8 экзонов. TATA-box на расстоянии около 30 нт.
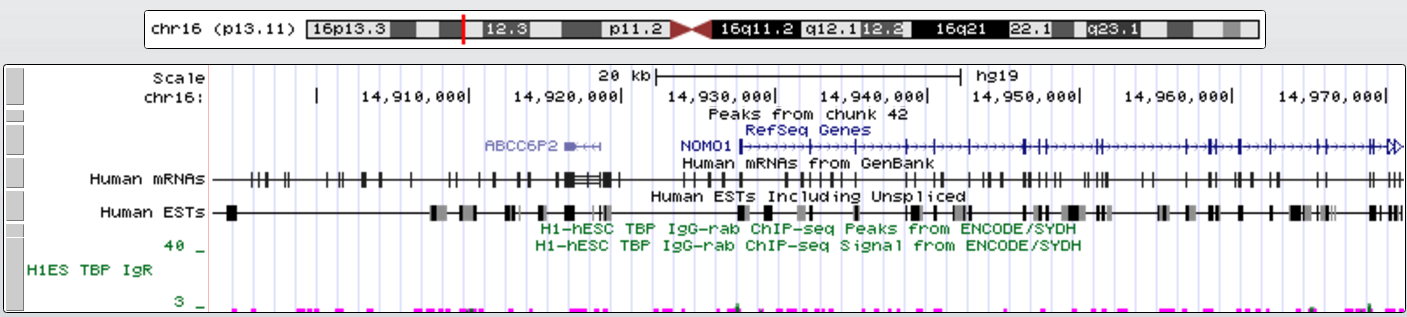
Пример гена без TATA-box - NOMO1 (NODAL modulator 1 - участвует в онтогенезе позвоночных). Его координаты chr16:14927643-14990014, длина 62372, + цепь, 31 экзон.
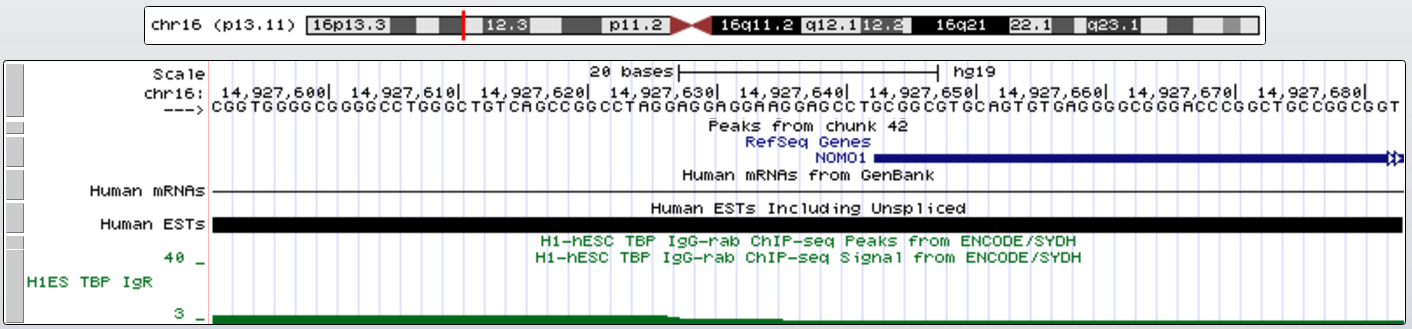
Во всех случаях TATA-box находился перед геном на расстоянии около 30-60 нт до старта транскрипции.