
Практикум 11
1. Описание файла с референсом
Файл chr8.fna похож на типичный файл в fasta-формате. Сразу после > идёт идентификатор RefSeq NC_000008.11, далее прописан источник последовательности (человек, 8 хромосома), затем идентификатор сборки генома GRCh38.p13 и статус сборки (первичная). Со следующей строки начинается запись последовательности ДНК. В начале и в конце большое количество неопознанных нуклеотидов.
2. Индексация референса
Для индексирования референсного генома использовалась команда bwa index -a bwtsw chr8.fna. На входе был файл с референсной последовательностью chr8.fna, а на выходе были получены файлы в форматах .amb, .ann, .bwt, .pac, .sa.
BWA — пакет программ, позволяющий картировать слабо расходящиеся с референстным геномом последовательности относительно этого генома. Команда bwa index индексирует референсный геном в fasta-формате. С помощью опции -a можно указать алгоритм для индексирования, в нашем случае это bwtsw.
3. Описание образца:
a. ссылка на информацию об образце;
b. прибор — Illumina Genome Analyzer IIx;
c. организм — Homo sapiens;
d. стратегия секвенирования — whole-exome;
e. парноконцевые чтения;
f. ожидаемое число чтений — 40990418 (41M spots).
4. Проверка качества исходных чтений
a. использованные команды: fastqc --help, fastqc SRR10720416_1.fastq.gz, fastqc SRR10720416_2.fastq.gz.
Отчёты: SRR10720416_1_fastqc.html, SRR10720416_2_fastqc.html.
b. количество пар чтений — 40990418;
c. количество прямых и обратных чтений совпадает;
d. количество чтений совпадает с ожидаемым (пункт 3f);
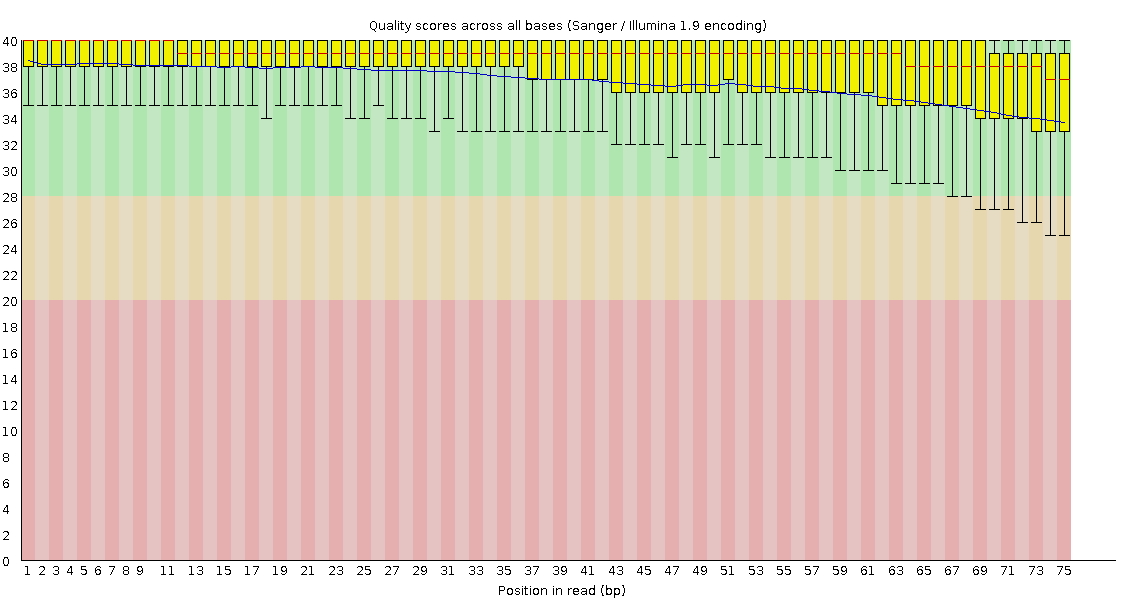
e. Per base sequence quality:
1. Прямые чтения
2. Обратные чтения
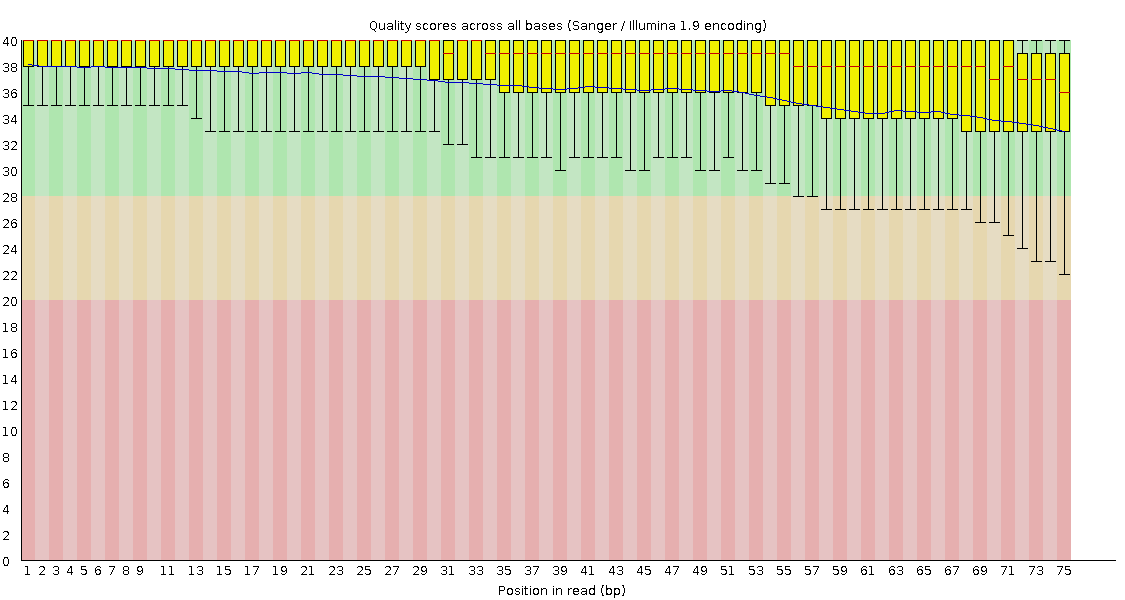
f. Чтения очень хорошие, Phred Quolity Score выше 20, то есть точность всегда больше 99%, большая часть лежит в зелёной зоне, но к самому концу точность падает и становится меньше 99,9%.
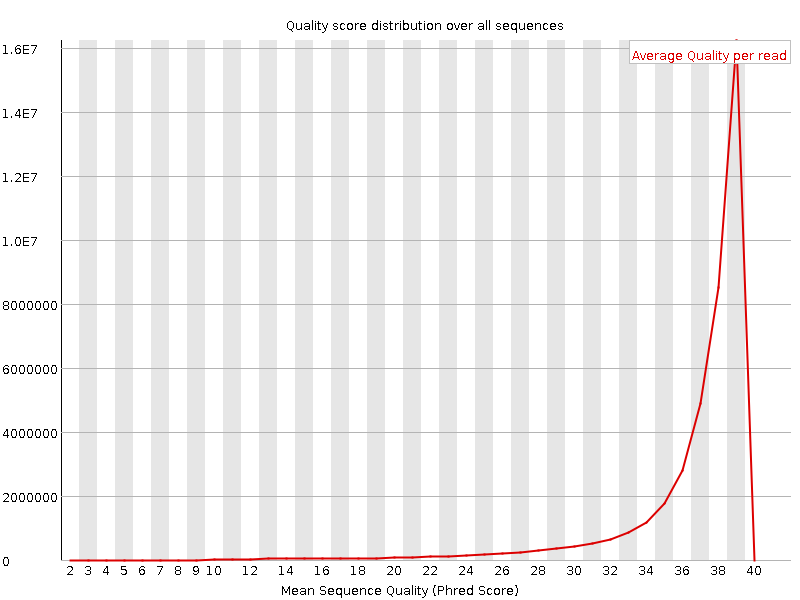
g. Per sequence quality scores:
1. Прямые чтения
2. Обратные чтения
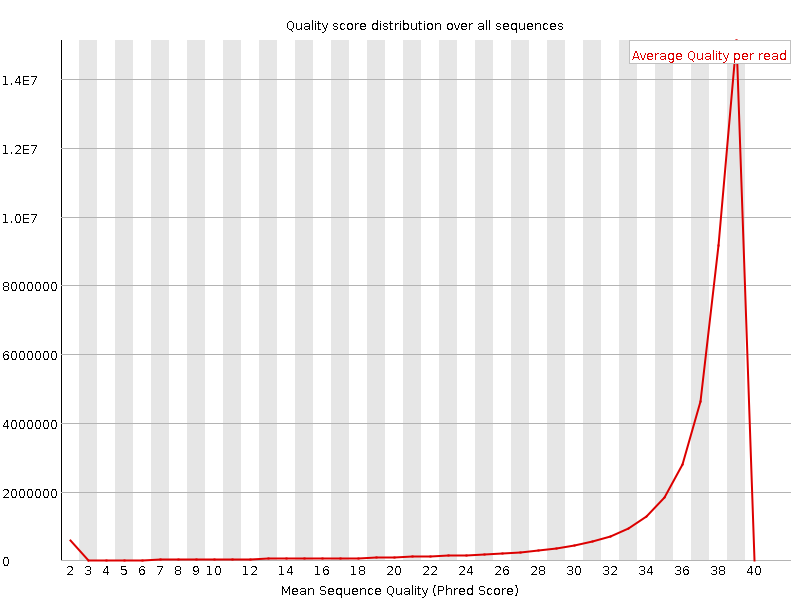
h. Графики показывают, что средний показатель качества (в обоих случаях) равен примерно 39, то есть точность очень высокая.
i. Sequence Length Distribution:
1. Прямые чтения
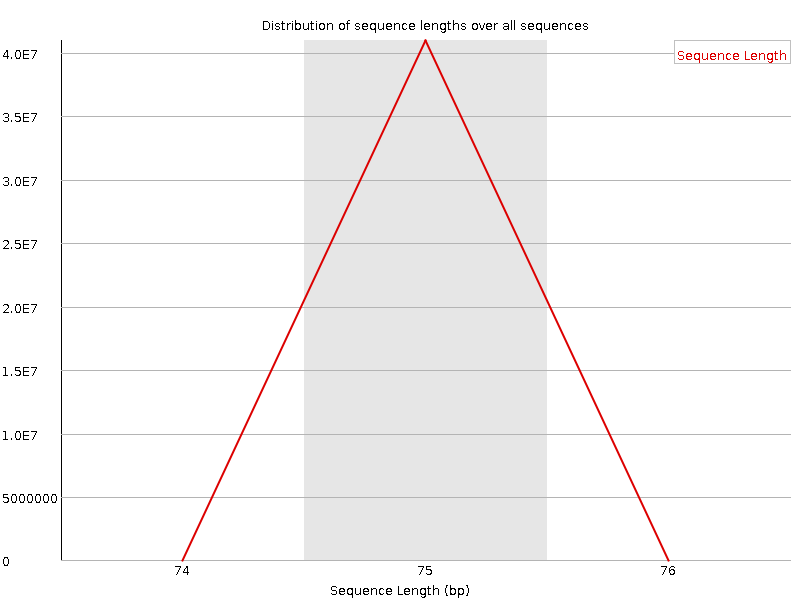
2. Обратные чтения
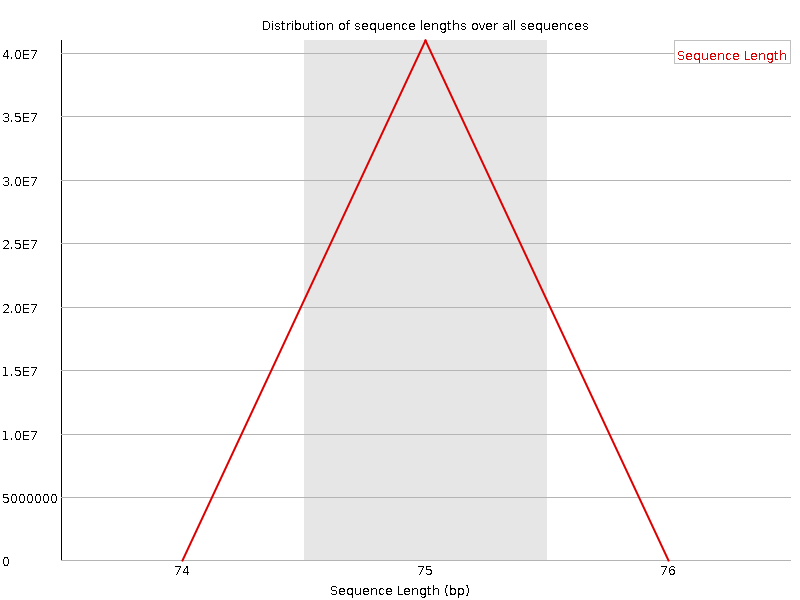
j. График показывает распределение длин фрагментов. Длины лежат в интервале от 74 до 76 с пиком в 75, что говорит о хорошей подготовке образцов перед секвенированием и помогает получить результаты с высоким качеством.
5. Фильтрация чтений
Для запуска программы Trimmomatic использовалась команда java -jar /usr/share/java/trimmomatic.jar PE -phred33 SRR10720416_1.fastq.gz SRR10720416_2.fastq.gz forward_paired.fastq.gz forward_unpaired.fastq.gz reverse_paired.fastq.gz reverse_unpaired.fastq.gz TRAILING:20 MINLEN:50.
- PE говорит о том, что работа идёт с парноконцевыми чтениями;
- -phred33 устанавливает Pred Quolity Score;
- следующие далее названия файлов задают вводные (первые два) и выходные (последние четыре) файлы;
- TRAILING:20 устанавливает порог качества, нуклиотиды с конца с качеством ниде будут удалены;
- MINLEN:50 удаляет риды с длиной меньше 50.
6. Проверка качества триммированных чтений
a. Для анализа полученных файлов с помощью программы fastQC использовались команды вида команды: fastqc file, где file соответствует названию исследуемых файлов.
Отчёты: forward_unpaired_fastqc.html, forward_paired_fastqc.html, reverse_unpaired_fastqc.html, reverse_paired_fastqc.html.
b. осталость 38977379 пар чтений (paired) это 95.09% от исходного количества;
c. Per base sequence quality:
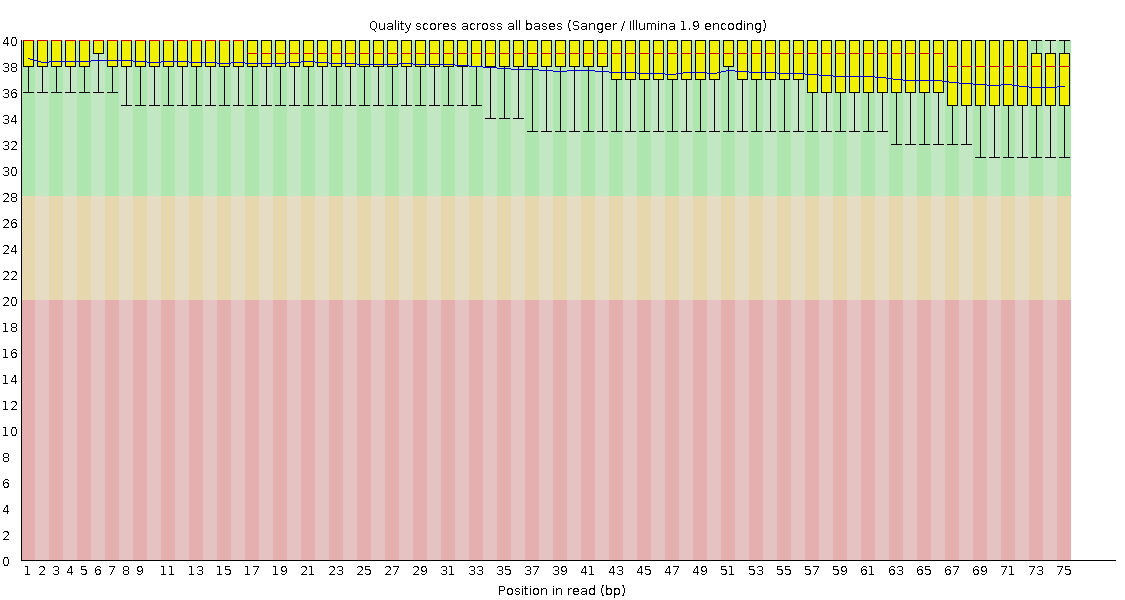
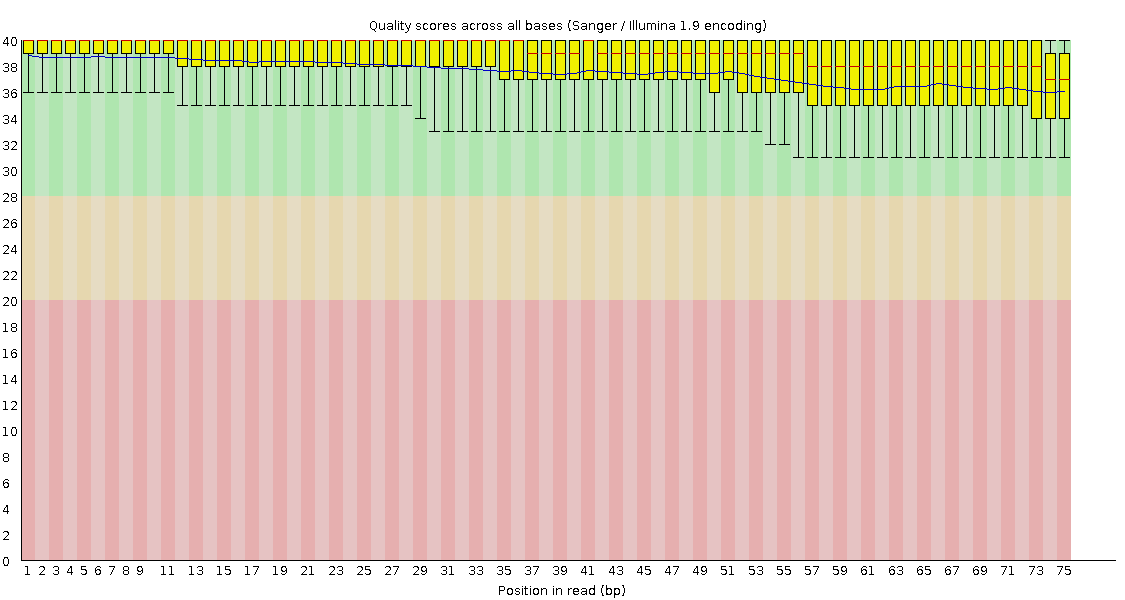
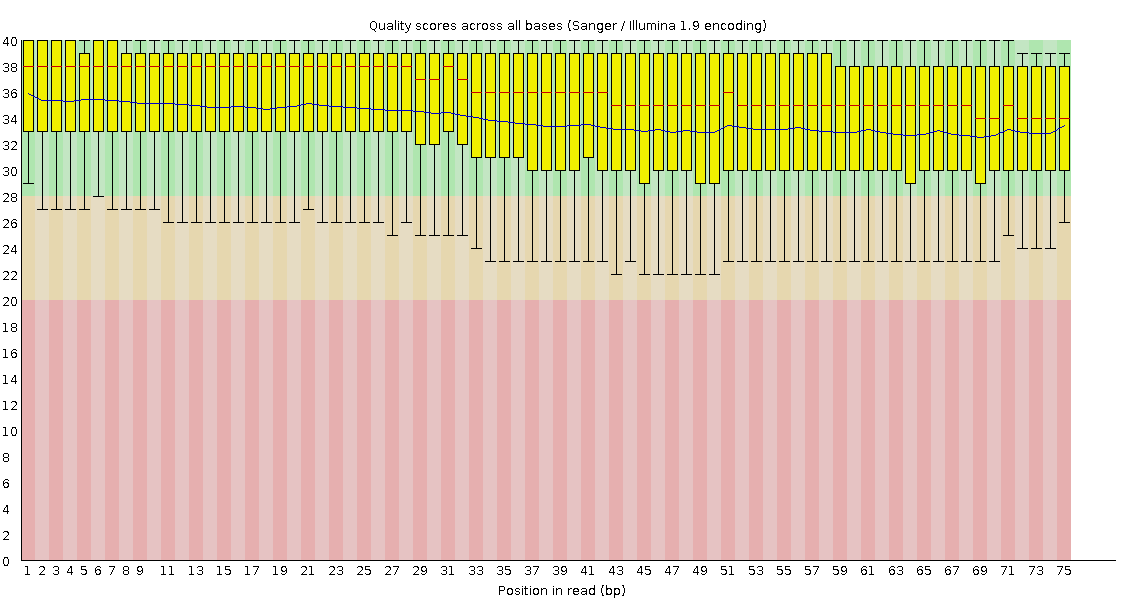
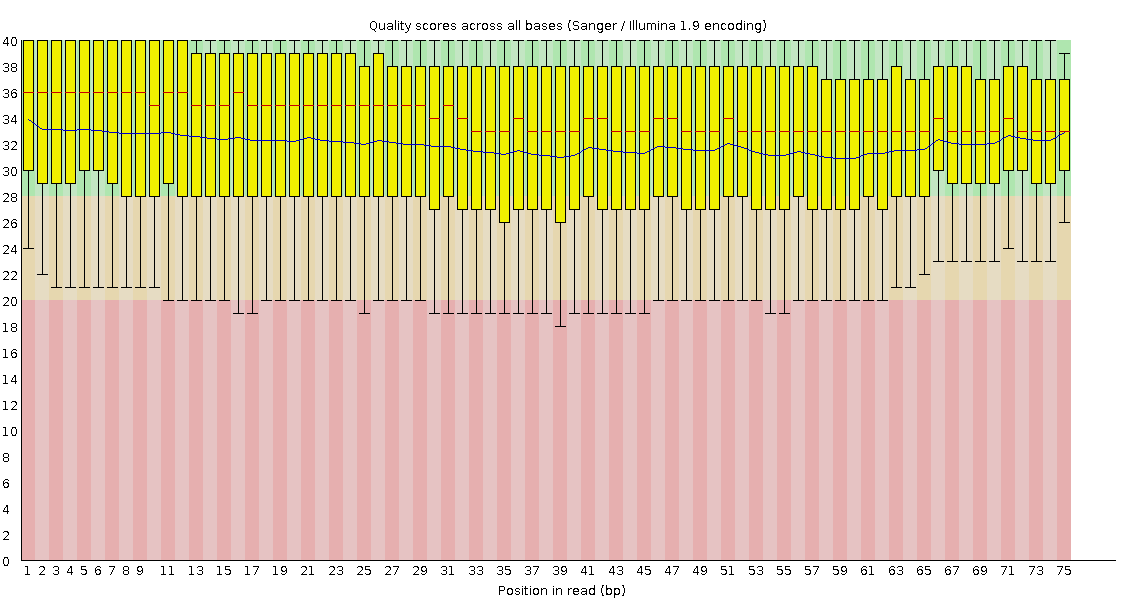
d. Сразу бросается в глаза, насколько хуже значения для unpaired по сравнению с paired. Для первых значения даже могут затрагивать красную зону, когда для последних значения даже не заходят в оранжевую. Таким образом, точность для unpaired ниже, чем для paired.
e. Paired очень близки к исходным чтениям, но являются более качественными.
f. Per sequence quality scores:
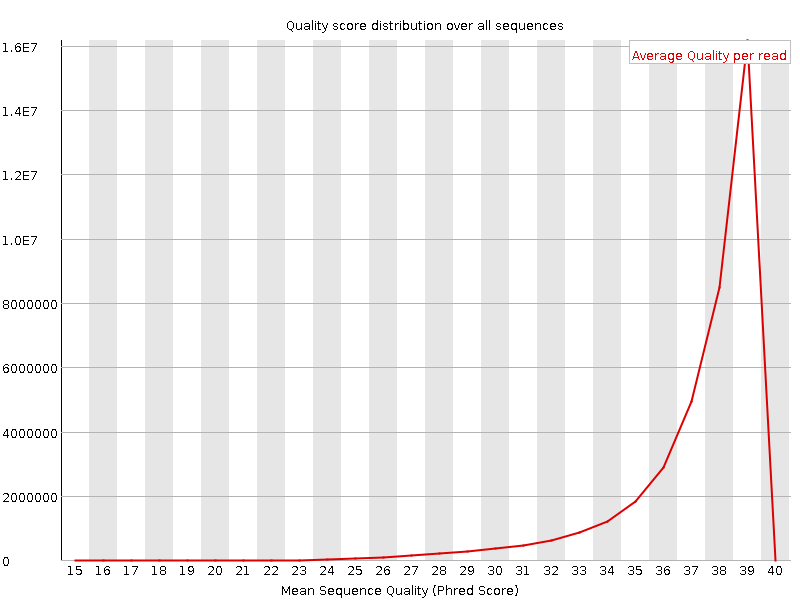
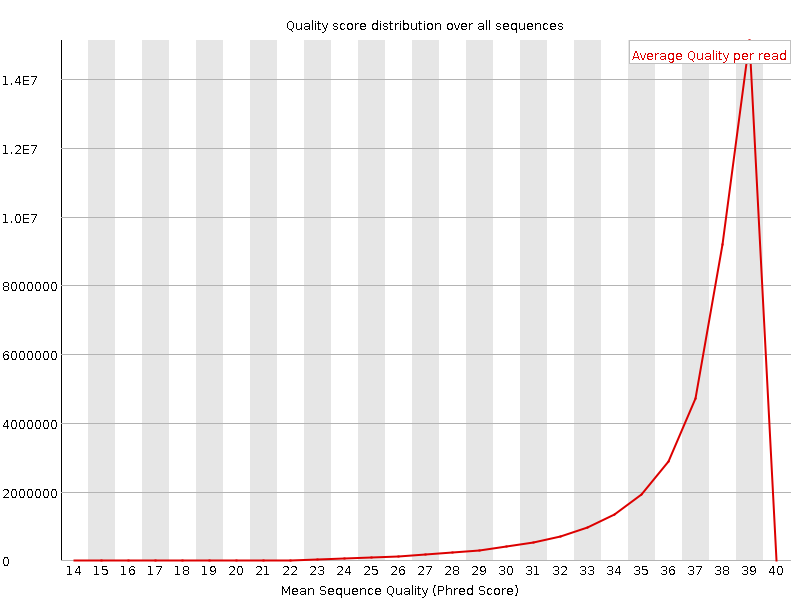
g. Заметных изменений по сравнению с исходными файлами нет, пик всё ещё приходится на 39.
h. Sequence Length Distribution:
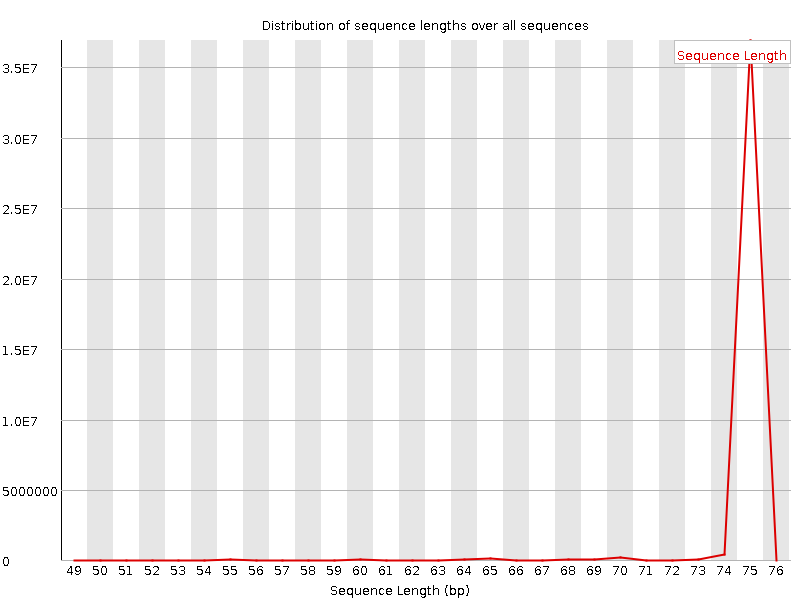
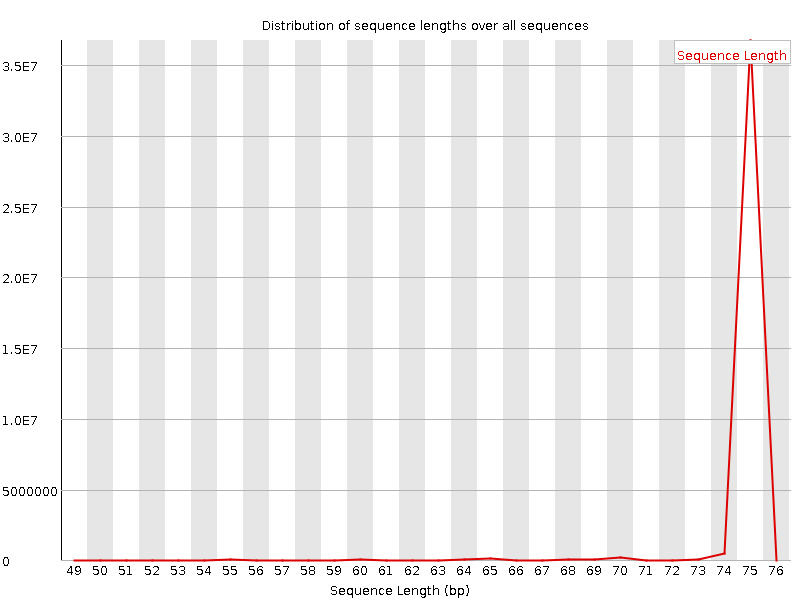
i. После триммирования основное распределение осталось тем же, но появились новые значения для длин меньше 74.