
Практикум 14
1. Подготовка референса
Проиндексируем референсный геном: hisat2-build chr8.fna chr8 2> hisat2.log.
- hisat2-build генерирует VCF-файл;
- chr8.fna — референс в формате fasta на входе;
- chr8 — префикс создающихся программой на выходе 8 бинарных .hr2 файлов;
- 2> hisat2.log — перенаправление логов в файл.
2. Оценка качества чтений
a. Проанализируем качество чтений с помощью fastqc SRR2015720_1.fastq.gz, результат можно посмотреть в файле SRR2015720_1_fastqc.html.
b. Всего получилось 30271388 чтений.
c. Per base sequence quality

d. Как видно из Изображения 1, чтения лежат в зелёной зоне, их качество достаточно хорошее. Ближе к концам происходит типичное уменьшение качества, но оно всё ещё лежит в приемлемом диапазоне.
Важно! В разделе Overrepresented sequences есть сообщение о часто повторяюще йся последовательности, похожей на адаптер "TruSeq Adapter, Index 2". Набор реактивов совпадает с указанным на странице записи. К сожалению, у меня так и не получилось привести файл с адаптерами к нужному виду для проверки с опциейй -c (файл не читался во время исполнения команды почему-то). Команда fastqc -c contaminant.txt --threads 10 SRR2015720_1.fastq.gz успехом не увенчалась. Но я всё же попробовал ужалить адаптеры командой java -jar /usr/share/java/trimmomatic.jar SE -phred33 -threads 10 SRR2015720_1.fastq.gz output.fastq.gz ILLUMINACLIP:adapt.txt:2:30:10, где adapt.txt — созданный мной fasta-файл с адаптером, который необходимо удалить (нашёл здесь). В итоге отпало 101662 (0.34%) ридов.
Далее я попробовал повторить анализ командой fastqc -t 10 output.fastq.gz, получил на выходе output_fastqc.html.
e. Sequence Length Distribution
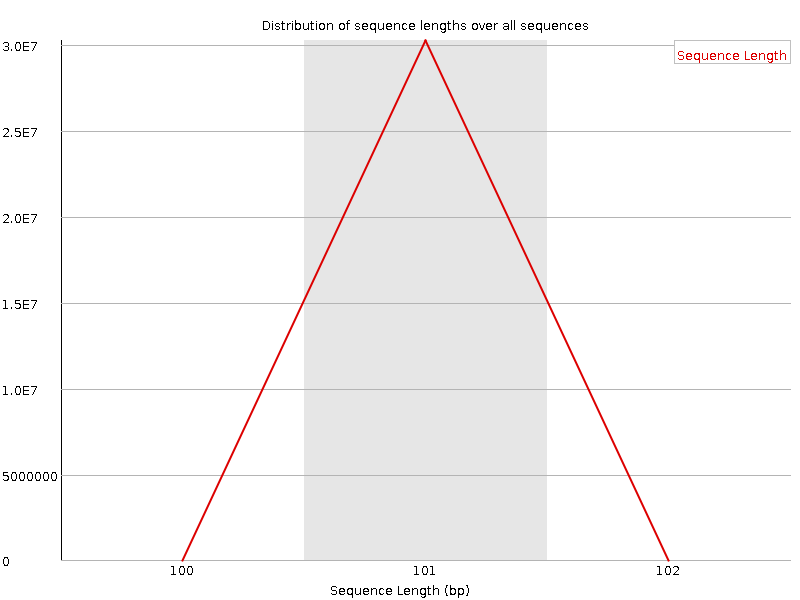

f. Как видно, после триммирования что-то пошло не так. До триммирования график прекрасен и понятен: длины ридов распределены между 100 и 102 с пиком в 101. Разбираться в причинах я не стал, продолжил работать с файлом до триммирования.
3. Картирование чтений на референс
a. Чтобы картировать NGS риды на наш геном, воспольщуемся командой hisat2 -p 10 -x ../chr8 -k 3 -U SRR2015720_1.fastq.gz > 14.sam 2> 14.log.
- -p 10 задаёт количество задействованных в процессе ядер;
- -x ../chr8 задаёт референс (chr8 — префикс созданных в п.1 файлов);
- -k 3 задаёт максимальное число выравниваний для каждого рида таких, что их score больше или равен значению score для всех остальных выравниваний;
- -U SRR2015720_1.fastq.gz задаёт список файлов с ридами. Имена файлов разделяются запятными без пробела, в нашем случае это одно название.
b. В файле 14.log найдём информацию о выравнивании чтений: всего из 30271388 наспаренных чтений 29215707 (96.51%) ни с чем не выровнялись (высокий процент объясняется картированием только на одну хромосому), 1034331 (3.42%) выровнялись 1 раз, 21350 (0.07%) больше одного раза.
c. Переведём 14.sam в bam формат:samtools sort -o 14.bam -O bam -@ 10 14.sam. Теперь проииндексируем: samtools index -@ 10 14.bam 14.bai.
d. Отберём чтения, что легли только на 8 хромосому: view -h 14.bam NC_000008.11 > chr8_14.sam. Конвертируем в .bam: samtools view -bS chr8_14.sam > chr8_14.bam.
4. Поиск экспрессирующихся генов
a. Рассмотрим файл с разметкой генов для 8 хромосомы. Он также состоит из шапки и тела. В шапке находится краткое описание, источник аннотации (проект GENCODE), контактный электронный адрес, формат и дата публикации. Тело содержит следующие стобцы:
- имя последовательности, в нашем случае хромосомы (NC_000008.11);
- source — источник аннотации (программа или база данных);
- feature — особенности гена, например, "CDS", "start_codon", "stop_codon, "exon";
- start — координата начала гена;
- end — координата конца;
- score — степень уверенности в координатах и самом существовании особеенности (feature), может (как в нашем случае) просто заменяться точкой, но некоторые программы указывают здесь в аннотации числа;
- strand — направление на цепи (на какой из цепей закодирован ген, + или -);
- frame — расстояние до первого кодона в нуклеотидных основаниях;
- attributes — информация об аннотации, обязательно содержит gene_id и transcript_id, может также указывать тип гена, уровень транскрипции, название и ID гена, прочие сведения;
- комментарии, начинаются с '#', не обязательно присутствуют в строке.
b. Можно попробовать получить количество "генов" по тегу с помощью команды awk '$3 == "gene"' gencode.chr8.gtf | wc -l (на выходе 2484), но, если просмотреть результат awk '$3 == "gene"' gencode.chr8.gtf | less, там окажутся элементы не только с пометкой "protein_coding", но и элементы с "processed_pseudogene", то есть псевдогены. Таким образом, выданное число превышает настоящее число генов.
c. htseq-count -f bam -s no -t exon 14.bam gencode.chr8.gtf > count.txt 2> count.log
- -f bam указывает формат файла с чтениями;
- -s no позволяет засчитывать совпадение, даже если чтение лежит на противоположнойцепи по сравнению с геном из аннотации;
- -t exon — значение в поле feature, в данном случае мы берём экзоны, т.к. исследовался экзом.
d. Файл count.txt был экспортирован в гугл-таблицы, чтобы легко посчитать количество чтений, попавших в границы генов. Так как значение опции -m я не указывал, оно по значению union, т.е, как видно на изображении 4 (взято из документации htseq-count), попавшие в границы генов чтения будут либо соответствовать одному конкретному гену, лиюо будут иметь значение __ambiguous, т.е. будут на пересечении генов.
В итоге в сумме получилось 797044 таких чтений. Вне границ генов (__no_feature) оказалось 237287 чтений.

e.
f.