
Предсказание вторичной структуры заданной тРНК
В этой части практикума продолжается работа с т-РНК 1G59, начатая в прошлый раз (страница практикума).
Предсказание через инвертированные повторы
Для предсказания вторичной структуры тРНК поиском инвертированных повторов использовалась команда einverted из пакета EMBOSS. В выдаче получились файлы 1g59.inv и 1g59.fasta.
Как бы я ни старался подогнать параметры штрафов и поощрений, адекватный результат выдавался только такой:
1G59: Score 15: 5/5 (100%) matches, 0 gaps
3 cccca 7
|||||
69 ggggt 65
Предполагаю, проблема в наличии достаточно большого количества неканонических взаимодействий в реальной структуре.
Предсказание вторичной структуры по алгоритму Зукера
Для предсказания использовалась программа RNAfold из пакета Viena Rna Package. Сначала необходимо было указать к ней путь: <export PATH=${PATH}:/home/preps/golovin/progs/bin>.
Далее использовался конвеер <cat 1G59.fasta | RNAfold --MEA>, выдача показана ниже, Изображение 1 в формате .ps (здесь уже конвертировано в png) сохранилось в рабочей папке без предупреждения об этом.
>1G59_1|Chains
GGCCCCAUCGUCUAGCGGUUAGGACGCGGCCCUCUCAAGGCCGAAACGGGGGUUCGAUUCCCCCUGGGGUCACCA
(((((((.(((((........)))))(((((.......)))))....(((((.......)))))))))))).... (-35.80)
(((((((.(((((........)))))(((((.......)))))....(((((.......)))))))))))).... [-36.21]
(((((((.(((((........)))))(((((.......)))))....(((((.......)))))))))))).... {-35.80 d=1.57}
(((((((.(((((........)))))(((((.......)))))....(((((.......)))))))))))).... {-35.80 MEA=72.59}
frequency of mfe structure in ensemble 0.511871; ensemble diversity 2.92
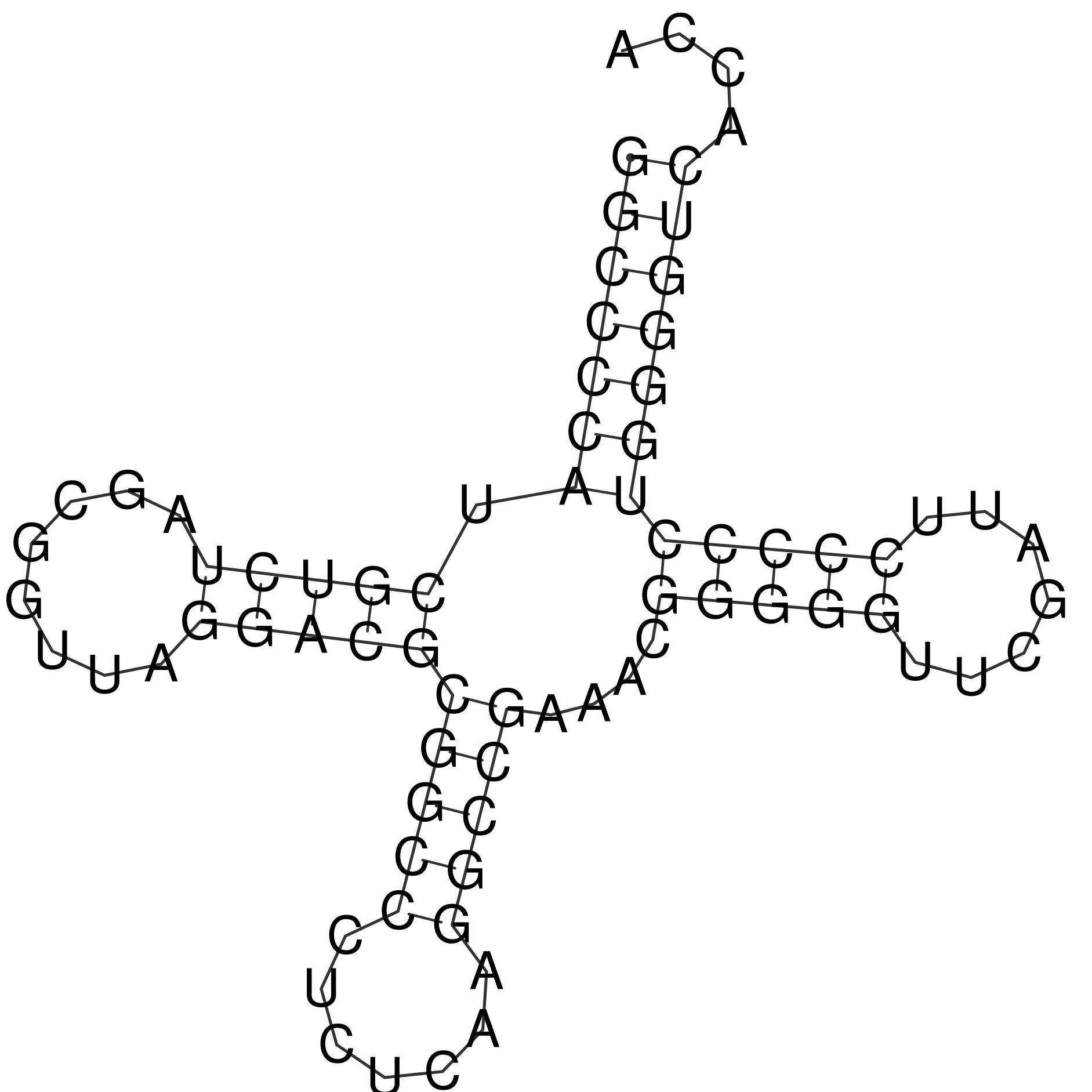
В Таблице 1 приводится сравнение трёх методов предсказания вторичной структуры тРНК. Как видно в таблице, программа einverted не дала необходимые результаты. Алгоритм Зукера хорошо предсказывает вторичную структуру, но необходимо отдельно подбирать параметры для идеального совпадения в каждом случае.
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
|---|---|---|---|
| Акцепторный стебель | 501–507/572–566 | предсказано 5 пар из 7 реальных | предсказано 7 пар из 7 реальных |
| D-стебель | 510–513/525–522 | - | предсказано 5 пар из 4 реальных |
| T-стебель | 549–553/565–561 | - | предсказано 5 пар из 5 реальных |
| Антикодоновый стебель | 538–544/532–526 | - | предсказано 5 пар из 7 реальных |
| Общее число канонических пар нуклеотидов | 20 | 5 | 18 |
Поиск ДНК-белковых контактов
Возвращение к работе в JMol
По заданию необходимо вспомнить, как задавать множества атомов. В апплете ниже скрипт сначала загружает структуру ДНК в проволочной модели, затем отображает на ней шарами последовательно множество атомов кислорода 2'-дезоксирибозы, множество атомов кислорода в остатке фосфорной кислоты и множество атомов азота в азотистых основаниях.
Поиск и характеристика ДНК-белковых контактов
В этой части практикума ведётся работа со структурой 1DDN.
За полярные принимались атомы кислорода и азота, за неполярные атомы углерода, фосфора и серы. При полярном контакте расстоение между полярным атомом белка и полярным атомом ДНК меньше 3.5Å, а при неполярном контакте неполярные атомы будут находиться на расстоянии меньше 4.5Å.
| Контакты атомов белка | Полярные | Неполярные | Всего |
|---|---|---|---|
| c остатками 2'-дезоксирибозы | 2 | 10 | 12 |
| c остатками фосфорной кислоты | 11 | 5 | 16 |
| c остатками азотистых оснований со стороны большой бороздки | 0 | 4 | 4 |
| c остатками азотистых оснований со стороны малой бороздки | 0 | 0 | 0 |
Можно заметить, что c остатками 2'-дезоксирибозы образуется больше неполярных контактов, а c остатками фосфорной кислоты больше полярных. Возможно, это объясняется процентным соотношением полярных и неполярных атомов в этих остатках.
Для обеих бородок не было найдено полярных контактов, неполярные были найдены только у большой бороздки. Скорее всего, это обусловлено труднодоступностью этих атомов цепи ДНК, особенно в малой бороздке.
Получение схемы ДНК-белковых контактов с помощью программы nucplot
Программа nucplot работает со старым форматом pdb-файлов, поэьтому сначала необходимо было конвертировать файл с помощью команды <remediator --old 1ddn.pdb > 1ddn_old.pdb>.
Далее с помощью команды <nucplot 1ddn_old.pdb> получили ps-файл с несколькими страницами схем ДНК-белковых контактов. Они представлены на Изображениях 1-4, на Изображении 5 изображён ключ к схемам.
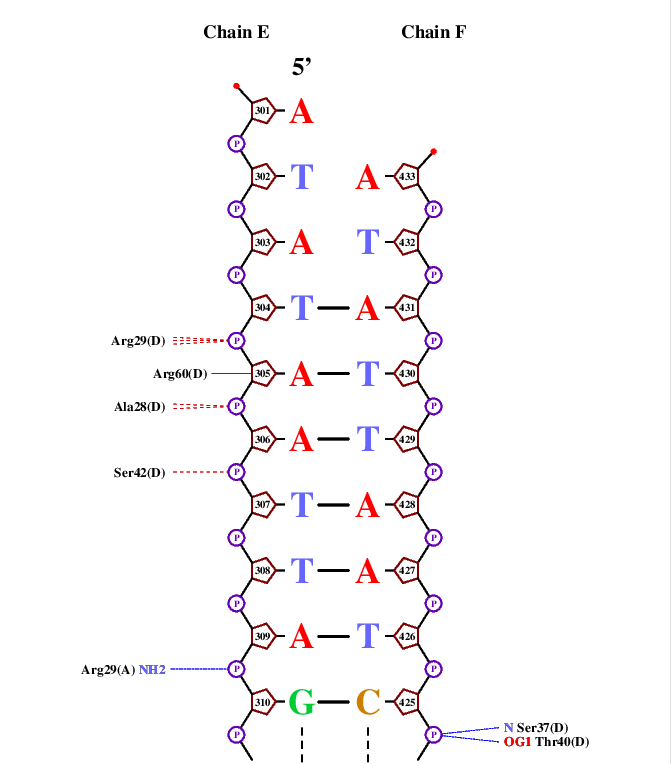
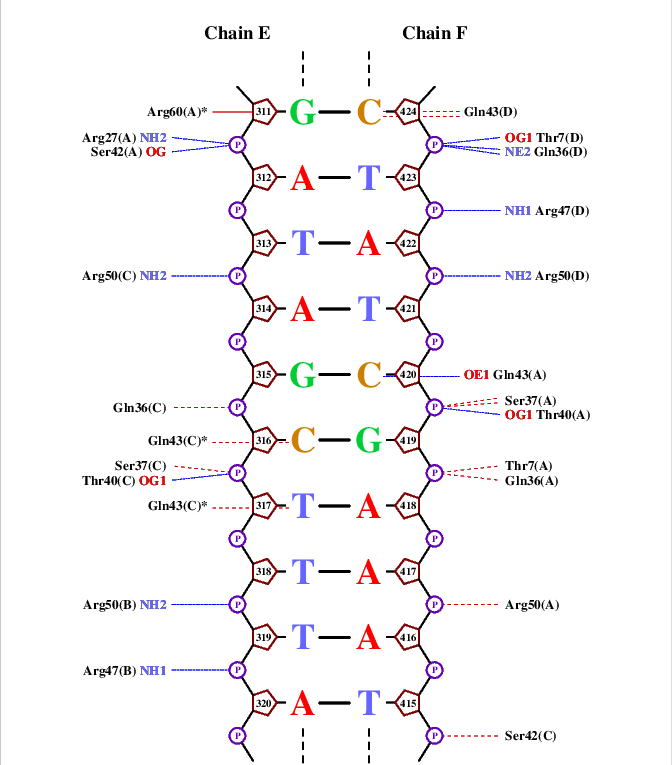
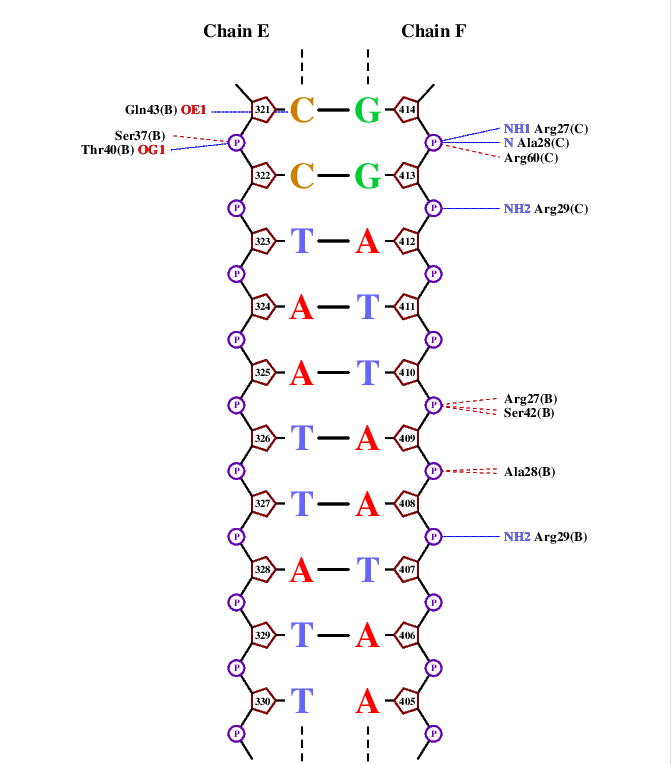


Наибольшее количество (а именно 3) контактов с ДНК у остатка Arg29(D). При этом, по видимому, эти контакты играют далеко не ведущую роль.
Можно предположить, что для узнавания и связывания с ДНК у каждой субъединице должны быть одинаковые контакты между ДНК и одними и теми же аминокислотными остатками, соответствующими симметрии субъединиц относительно ДНК (A/C, B/D). Если следовть такой логике, самыми важными остатками окажутся:
- Arg29, взаимодействующий с атомами фосфорного остатка
- Thr40, образующий сильное взаимодейтсвие с кислороом OG1 цитозина
- Arg50, связь с фосфорным остатком в симметрии A/B, C/D (