



1. Получение консенсусной последовательности на основании данных из капиллярного секвенатора
Работа велась с выданными капиллярным секвенатором файлами 35_F.ab1 и 35_R.ab1 для прямой и обратной цепи соответственно. В них уже есть автоматическая аннотация нуклеотидов (base calling), но очень часто в ней содержатся ошибки.
Для просмотра хроматограмм использовалась программа UGENE. На всякий случай на будущее приведу для себя команды для запуска (иначе я потом не вспомню, в какой операционной системе и где что лежит):
cd Загрузки tar -xzf ugene-37.0-linux-x86-64.tar.gz #распаковка скачанного пакета cd ugene-37.0 ./ugeneui
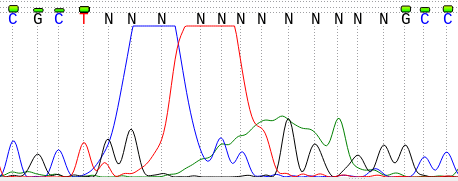
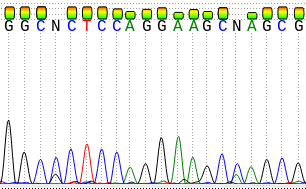
Сначала просмотрим обе хроматограммы. В целом качество хорошее, шума мало. Однако хроматограмма для обратной цепи похуже, в ней есть пятно краски (Рис.1), в нескольких местах сигнал настолько слабый, что его можно спутать с шумом. Средняя сила сигнала одинакова на всей последовательности.
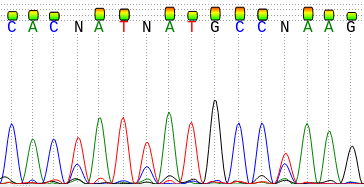
Также сразу бросается в глаза количество полиморфизмов (сила второго сигнала в той же позиции больше 2/3 первого или равна силе первого), но их наличие нужно будет проверить в ходе выравнивания прямой и обратной последовательности. Пример приведёт на Рисунке 2.
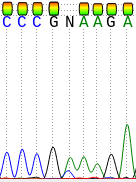
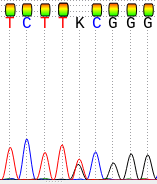
В ходе анализа встречалось множество проблемных нуклеотидов и полиморфизмов, приведу несколько примеров. На Рис.3 изображён проблемный нуклеотид в прямой цепи. Сигнал цитозина слишком маленький, чтобы без сомнений поставить M, но в то же время слишком большой, чтобы однозначно отнести его к шуму. Проверим комплементарный нуклеотид в обратной цепи (Рис.4). Сигналы для T и G одинаковые, видим полиморфизм. Следовательно, в прямой цепи имеем A или C, ставим M в соответствии с кодом вырожденных нуклеотидов.
Ещё один участок, который необходимо было восстановить по комплементарной цепи (из-за пятна), изображён на Рисунке 1, комплементарный участок на Рисунке 5, т.о. восстанавливаем 14 нуклеотидов.
Последовательности были скопированы в файлы reverse_1.fa и forward_red.fasta, чтобы можно было спокойно выделять строчными буквами замены и заменять нуклеотиды на обозначения из кода вырожденных нуклеотидов (UGENE так не умеет). Затем последовательности были скопированы в reverse_1.fasta, forward_1.fasta и переведены в верхний регистр. Это нужно для выравнивания. Для начала получим обратную последовательность для обратной цепи командой revseq reverse_1.fasta rev_1.fasta. Затем выровняем последовательности: needle forward_1.fasta rev_1.fasta -aformat3 fasta -out alignment_1.fasta, штрафы за гэпы брались дефолтные.
Далее выравнивание alignment_1.fasta (уже без нечитаемых концов) визуализировалось в JalView (Рисунок 6). Это нужно, чтобы увидеть оставшиеся проблемные участки. Нечитаемые концы в координатах выравнивания было нелогичено брать, потому что неопределённные нуклеотиды имели в нём нулевое покрытие. В количестве аннотиванных нуклеотидов в хроматограмме для прямой и обратной цепи длины нечитаемых участков составляют 35 и 19 нуклеотидов соответственно.

Консенсусная последовательность копировалась непосредственно из JalView.
2. Нечитаемый фрагмент хроматограммы
На Рис.1 уже изображён нечитаемый фрагмент. Это пятно от краски, техническая ошибка.
