Практикум 2. Реконструкция филогении. Просто описание процесса, чтобы самому потом вспомнить...
Выбранные бактерии
| № | Название | Мнемоника |
|---|---|---|
| 1 | Agrobacterium fabrum | AGRFC |
| 2 | Brucella suis | BRUSU |
| 3 | Burkholderia cenocepacia | BURCA |
| 4 | Haemophilus influenzae | HAEIN |
| 5 | Rhizobium meliloti | RHIME |
| 6 | Roseobacter denitrificans | ROSDO |
| 7 | Saccharophagus degradans | SACD2 |
| 8 | Shewanella denitrificans | SHEDO |
Выравнивание последовательностей фактора элонгации трансляции Ts (мнемоника EFTS)
Открыл JalView командой jalview в терминале, в меню File выбрал Fetch sequences, в окне Select Database выбрал Uniprot. Убрал галочку у Autosearch, а то мешает тратой времени на загрузку после каждого редактирования запроса. Вбил запрос "EFTS_AGRFC; EFTS_BRUSU; EFTS_BURCA; EFTS_HAEIN; EFTS_RHIME; EFTS_ROSDO; EFTS_SACD2; EFTS_SHEDO", где на первом месте мнемоника белка, а на втором после _ идёт мнемоника организма из 1 практикума. Выделил выдачу, нажал Ok внизу. Открылось окно с последовательностями. В меню выбрал Web Service → Alignment → Run Muscle With Defaults. Получилось выравнивание. Сохранил его в формате fasta, переписал в файле для удобства аннотации на мнемоники организмов.
Открыл MEGA. File → Open a File/Session. Появилось всплывающее окно "Analyze or align?". Выбрал Analize, в новом окне выбрал Protein sequences.
Реконструкция филогении тремя методами
Файлы в папке Документы → Alignment.
Maximum Likelihood method, Evolutionary relationships UPGMA
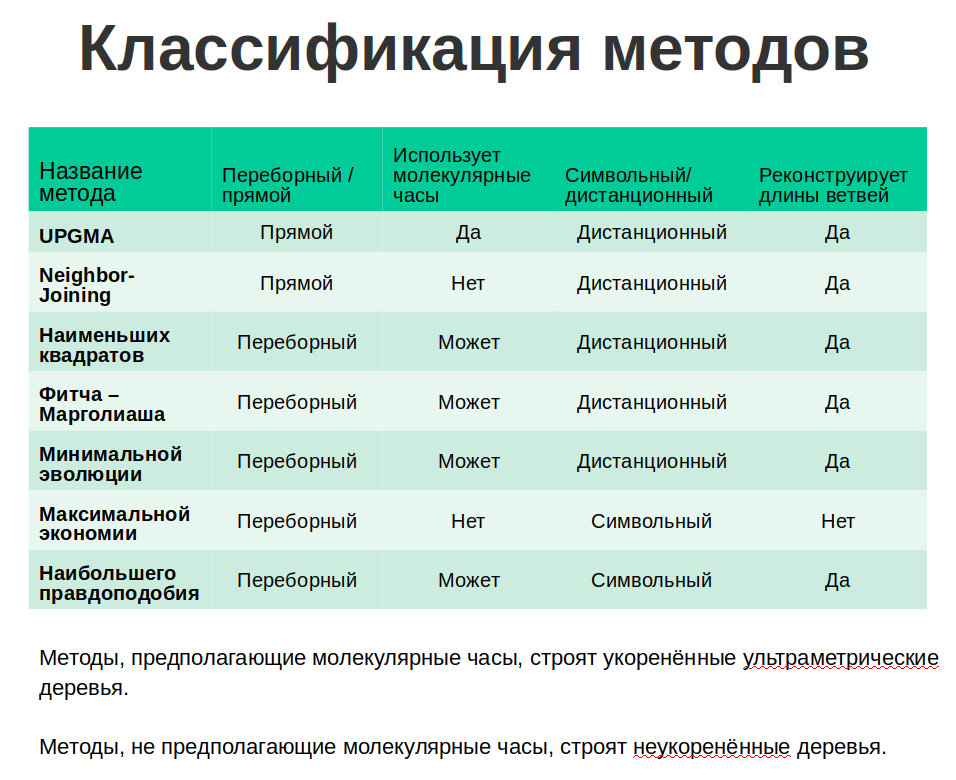
UPGMA = «Unweighted pair group method with arithmetic mean» Строит укоренённое ультраметрическое дерево. Возможно, лучший из методов, предполагающих молекулярные часы.
Все методы, кроме максимальной экономии, допускают предположение о молекулярных часах (но чаще используются без этого предположения!) и оценивают длины ветвей.
Методы MP и ML — символьно-ориентированные, LS, FM, ME и многие другие принимают на вход матрицу расстояний.