|
|
Одноконцевые RNA-Seq чтения из биологической реплики 1
(файл с чтениями chr8.1.fastq) картировались
на референсную последовательность хромосомы 8 H. sapiens.
На основании разметки человеческого генома Gencode 19 для сборки hg19
определялись гены, с которых были транскрибированы отсеквенированные РНК
выбранной реплики.
|
Методы
Все полученные файлы хранятся в рабочей директории
/nfs/srv/databases/ngs/stepan_puhov/pr12
в соответствующих поддиректориях.
Далее представлена таблица, отражающая последовательность действий и
использованных команд (для точности описания указывается, в каких директориях
производился запуск соответствующих команд):
| Действие | Команда |
| Первичный контроль качества чтений | (в директории reads)
fastqc --extract chr8.1.fastq |
| Попытка очистить чтения | (в директории reads)
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/ trimmomatic-0.30.jar SE -phred33 chr8.1.fastq chr8_trimmed.1.fastq TRAILING:20 MINLEN:25 |
| Картирование чтений на референсную последовательность | (в директории alignments)
hisat2 --no-softclip --summary-file mapping_report.txt -x ../../pr11/reference/chr8 -U ../reads/chr8.1.fastq -S chr8.sam |
| Подсчёт чтений, картированных по принципу spliced-alignment | (в директории alignments)
grep -E -c "^[^@]([A-Za-z0-9:]+\s){5}[0-9A-Z]+[0-9]+N" chr8.sam |
| Перевод картированных чтений в bam формат | (в директории alignments)
samtools view -b -o chr8.bam chr8.sam |
| Сортировка bam-файла по координатам картирования чтений | (в директории alignments)
samtools sort chr8.bam chr8_sorted |
| Индексирование отсортированного bam-файла (создаётся файл chr8_sorted.bam.bai) | (в директории alignments)
samtools index chr8_sorted.bam |
| Подсчёт картированных чтений, попадающих на гены, способом "union" | (в директории alignments)
htseq-count -f bam -s no chr8_sorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/*.gtf > reads_count_union.table |
| Подсчёт картированных чтений, попадающих на гены, способом "intersection-strict" | (в директории alignments)
htseq-count -f bam -s no -m intersection-strict chr8_sorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/*.gtf > reads_count_intersectstrict.table |
| Поиск непустых features в выдаче программы htseq-count | (в директории alignments)
grep -E -e "^[A-Z0-9\.]+\s[^0]" -e "^__" reads_count*.table |
|
Анализ качества чтений
С помощью программы FastQC был получен
отчёт о качестве чтений.
Из раздела отчёта "Basic statistics" видно, что в исследуемом образце всего
17763 чтений, длина чтений составляет от 25 nt до 51 nt, ни одно чтение не
помечено как "чтение плохого качества" и используется кодировка качества
Phred+33.
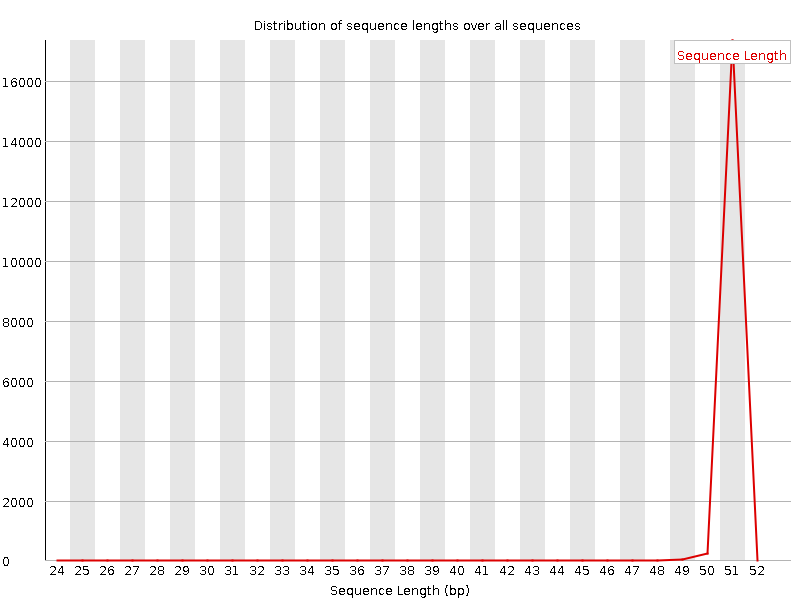
Из раздела отчёта "Sequence length distribution" понятно, что большинство
чтений имеют длину более 49 nt:
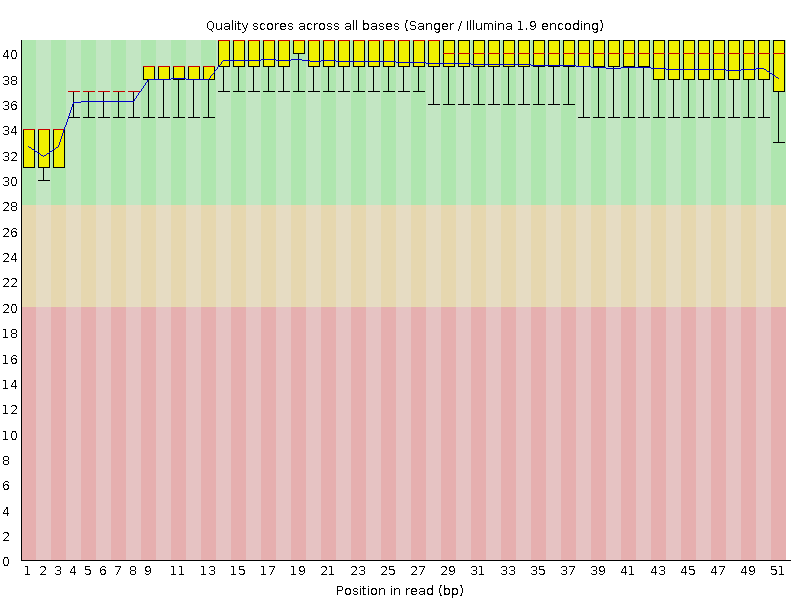
В разделе отчёта "Per base sequence quality" видим, что большинство чтений
имеют очень хорошее качество; для каждого выбранного нуклеотида более, чем в
90% случаев, качество будет хорошим (Q > 28):
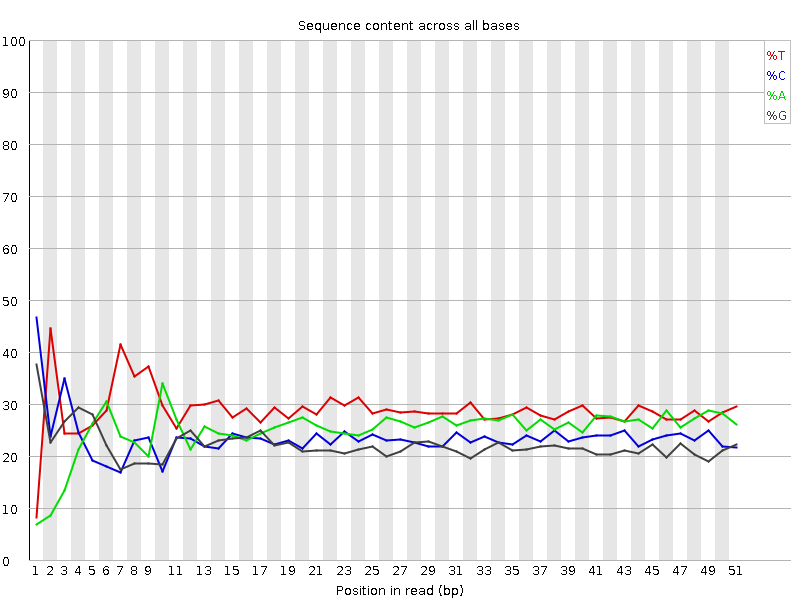
В разделе отчёта "Per base sequence content" выдаётся ошибка, ибо наблюдается
картина сильно отклоняющаяся от того, что ожидается от случайной библиотеки
чтений, полученных с одного генома, где частота каждого основания должна быть
примерно одинаковой в любой позиции чтения (примерно равной частоте в геноме):
Однако это не удивительно для библиотеки секвенирования РНК из-за
перепредставленности некоторых чтений, связанной с разными уровнями экспрессии
генов, с которых считываются секвенированные транскрипты − частота
оснований в каждой позиции, таким образом, смещена в сторону средней частоты
по соответствующей позиции в РНК генов с высокой экспрессией.
По той же причине разного содержания разных РНК в исследуемом образце выдаётся
ошибка в разделе отчёта "Sequence duplication level", из которого видно, что
при удалении копий повторяющихся чтений остаётся лишь 36.93% чтений, и
предупреждение в разделе "Overrepresented sequences" − это никак не
говорит о качестве чтений.
В целом, можно утверждать, что чтения имеют очень хорошее качество.
Попытка очистить чтения программой Trimmomatic, отрезав с 3'-конца
нуклеотиды с Q < 20 и при этом сохранив минимальную длину чтений равной 25 nt,
оказалась бесполезной − ни одно чтение не было триммировано.
|
Картирование чтений и анализ выравнивания
Картирование чтений на референсную последовательность было произведено
программой hisat2 c отключенной
опцией --no-spliced-alignment.
Отключение этой опции позволяет программе разделять чтение на участки и
картировать полученные участки независимо на одну и ту же цепь референса в
предположении о наличии "сплайсинг-сайта" (вырезанного интрона) в данном
чтении. Для чтения, картированного таким способом, в поле CIGAR sam-файла
указывается длина прерывающего его интрона (кодом в регулярном выражении
[0-9]+N); с помощью программы grep было подсчитанно, что так
картировалось 4514 чтений.
Из отчёта картирования программы
hisat2 видно, что из 17763 данных программе чтений 304 чтения не
картировались, а из остальных чтений 17455 картировались ровно 1 раз и
4 чтения картировались более 1 раза. Таким образом, всего картировалось
98.29% чтений.
С помощью программы grep было выяснено, что у всех 17455 чтений, что
картировались лишь 1 раз, было высокое качество картирования (Q = 60), а
у 4 чтений, которые картировались более 1 раза, качество картирования было
очень низким (Q = 1 или Q = 0). Можно заключить, что картирование, в целом,
имеет высокое качество.
Для дальнейшей работы файл sam-формата с картированными чтениями с помощью
программ пакета Samtools был переведён в bam-формат, а полученный
bam-файл был отсортирован по координате картирования чтений и проиндексирован.
|
Подсчёт чтений
Подсчёт чтений, картированных на гены, определяемые согласно разметке
Gencode 19 сборки hg19, производился программой htseq-count.
Программа принимает файл с картированными чтениями и файл формата gtf или gff
с разметкой генома и для каждого участка генома, выделенного данной разметкой,
(так называемого feature) считает число попавших на него чтений по 1 из 3
алгоритмов: union (для каждого чтения учитывается вхождение в участок генома
(feature) хотя бы 1 нуклеотидом), intersection-strict (для каждого чтения
учитывается вхождения в участок генома (feature) только по всем нуклеотидам
чтения) или intersection-nonempty (для каждого чтения учитывется вхождение
в feature по всем нуклеотидам чтения, входящих хотя бы в
какой-нибудь feature).
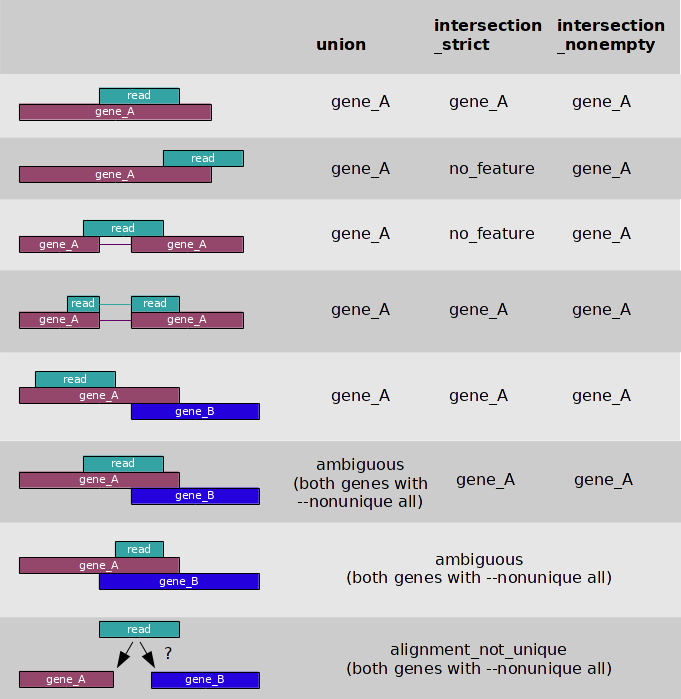
В данном случае за features принимались гены, как совокупность их экзонов
(то есть вхождение чтения в feature по данному нуклеотиду − это вхождение
этого нуклеотида чтения в какой-нибудь экзон данного гена). При этом 3
алгоритма подсчёта чтений могут давать следующие результаты в разных ситуациях:
Важные опции для запуска htseq-count:
-
-f определяет формат входного файла с картированными чтениями
-
-s значение "yes" считает чтения для feature, только если они на той же цепи,
что и feature, значение "reverse" − только если чтения на противоположной
цепи, значение "no" позволяет считать чтения, картирующиеся на обе цепи по
отношению к данному feature
-
-t определяет название для выбранного типа feature во входном gtf файле
(default: exon)
-
-i определяет аттрибут в записях gtf файла, значение которого принимается
за feature ID (default: gene_id)
-
-m определяет 1 из 3 способов подсчёта чтений (default: union)
Сначала был проведён подсчёт чтений, ложащихся на гены, представленные как
совокупности экзонов, (то есть определние генов дефолтными значениями опций
-t -i) по рекомендованному алгоритму union:
ENSG00000104738.12 294
ENSG00000253729.3 15941
__no_feature 1219
__ambiguous 1
__too_low_aQual 0
__not_aligned 304
__alignment_not_unique 8
Затем чтения, ложащиеся на гены (при том же определении генов), были подсчитаны
по алгоритму intersection-strict:
ENSG00000104738.12 273
ENSG00000253729.3 15511
__no_feature 1671
__ambiguous 0
__too_low_aQual 0
__not_aligned 304
__alignment_not_unique 8
Из вывода программы htseq-count выясняется, что
-
304 чтения не были картированы
(они были определены ещё программой hisat2);
-
8 выравниваний не были
уникальными (это выравнивания для тех самых 4 чтений с более, чем 1
картированием; у каждого из них есть 2 координаты картирования, что видно из
sam файла);
-
1 чтение частично лежит на пересечении экзонов 2 разных генов;
-
1671 чтение хотя бы частично попадают на интроны, а частично могут попадать
на экзоны гена PRKDC или гена MCM4, из них 1219 чтений полностью попадают
на интроны;
-
273 и 15511 чтений полностью попадают на экзоны генов MCM4 и PRKDC
соответсвенно.
Вообще, для чтений, полученных секвенированием мРНК, ожидаются
картирования, как на строках 1 или 4 приведённой выше в этом разделе схемы.
Картирования полностью в интрон или одним концом в интрон можно об'яснить
присутствием в чтениях некодирующих РНК (а таких, например, 2 в гене PRKDC) или
контаминацией незрелыми мРНК из ядра, а картирование на 2 экзона и интрон
между ними (как в строке 3 схемы) об'ясняется скорее всего ядерной
контаминацией незрелыми мРНК. Случай из 6 строки схемы, с пересекающимися
экзонами с разных генов не понятен (а такой присутствует в 1 из исследуемых
чтений, судя по разному подсчёту 2 разными алгоритмами поля "ambiguous").
В результате, чтения, картированные на экзоны, попали на 2 соседних гена:
-
PRKDC − ген каталитической суб'единицы ДНК-активируемой протеинкиназы,
серин/треониновой протеинкиназы, участвующей в NHEJ-репарации.
-
MCM4 − один из 8 паралогичных генов, белки которых образуют
гетерооктамерный комплекс, выполняющий роль главной хеликазы при репликации
ядерной ДНК.
|
|
|