Электронная плотность. Рассеяние электрона
Выбор структуры
Структура белка для дальнейшей работы должна удовлетворять некоторым требованиям. Был взят белок из первых семестров (YABJ_BACSU, 1QD9) и проверен по следующим критериям:
- В PDB выложены экпериментальные данные о структуре, а не только модель. Доступен для скачивания файл структурных факторов (экспериментальные данные), EDS узнает PDB ID и предоставляет всевозможную полезную информацию о структуре.
- В PDBeFold находится множество разнообразных гомологов белка разного сходства из разных организмов. Минимальное RMSD составляет 0.62. Для дальнейшей работы были выбраны несколько белков из разных организмов с разным RMSD и числом сопоставленных Cα:
Name RMSD N_align (total 124) Length PDB ID CRYSTAL STRUCTURE OF PUTATIVE TRANSLATION INITIATION INHIBITOR PH0854 FROM PYROCOCCUS HORIKOSHII 0.85 119 125 2dyy CRYSTAL STRUCTURE OF E. COLI TDCF WITH BOUND SERINE 1.00 122 127 2uyk CRYSTAL STRUCTURE OF A PUTATIVE TRANSLATION INITIATION INHIBITOR FROM SALMONELLA TYPHIMURIUM 1.66 109 115 3gtz CRYSTAL STRUCTURE OF A PUTATIVE ENDORIBONUCLEASE (SO_1960) FROM SHEWANELLA ONEIDENSIS MR-1 AT 1.85 A RESOLUTION 2.05 106 153 1otm CRYSTAL STRUCTURE OF A PROTEIN OF UNKNOWN FUNCTION PA3499 FROM PSEUDOMONAS AERUGINOSA 1.55 117 142 1ig8
Файл структурных факторов
Из PDB был загружен файл структурных факторов и сверен непосредственно с файлом структуры pdb. Видно, что в файле структурных факторов число записей не соответствует количеству атомов в структуре. Собственно, в самом файле и описано, что за информация в нем содержится:
loop_ _refln.crystal_id _refln.wavelength_id _refln.scale_group_code _refln.index_h _refln.index_k _refln.index_l _refln.status _refln.F_meas_au _refln.F_meas_sigma_au 1 1 1 0 0 18 o 512.900 19.800 1 1 1 0 0 24 o 1435.700 60.100 1 1 1 0 0 30 o 1577.800 66.700 ...
Каждая строчка _refln. соответствует столбцу данных. Поля соответствуют mmCIF формату. Таким образом, в файле для каждой тройки Миллеровых индексов h, k и l указаны модуль и стандартное отклонение структурного фактора. Его фазу, как известно, детектировать нельзя. Кроме того, указаны ID кристалла, длины волны возбуждения и код группы масштабирования (в этой структуре они одинаковы для всех записей, ячейка только одна, длина волны составляет 1.0095 А), а также статус, который влияет на участие этого рефлекса в дальнейших рассчетах. Это можно считать сырыми данными, по которым потом можно восстановить структуру в декартовых системах координат и записать ее в pdb файл. Кроме того, в файле можно найти и сведения о размере и углах ячейки.
Электронная плотность
С сайта EPS был скачан файл электронной плотности структуры. С помощью pymol он был наложен на саму структуру, чтобы оценить качество ее модели. На рисунках показаны различные уровни электронной плотности на всей полипептидной цепи без боковых радикалов. Учитывалась электронная плотность только на расстоянии, меньшем или равном 1.9 А от молекулы (так отсеклись все "лишние", то есть соответствующие соседним молекулам, электронные плотности и шум). Использовались уровни электронной плотности, равные 1, 2 и 3 σ. Последний порог был слишком велик для ее адекватного отображения вокруг полипептидной цепи, на двух сигмах тоже видны "пропадающие" участки структуры. При пороге в одну сигму появляется очень много шума (что естественно), но на картинках он был по большей части обрезан, так как не вошел в область 1.9 А от полипептидной цепи.
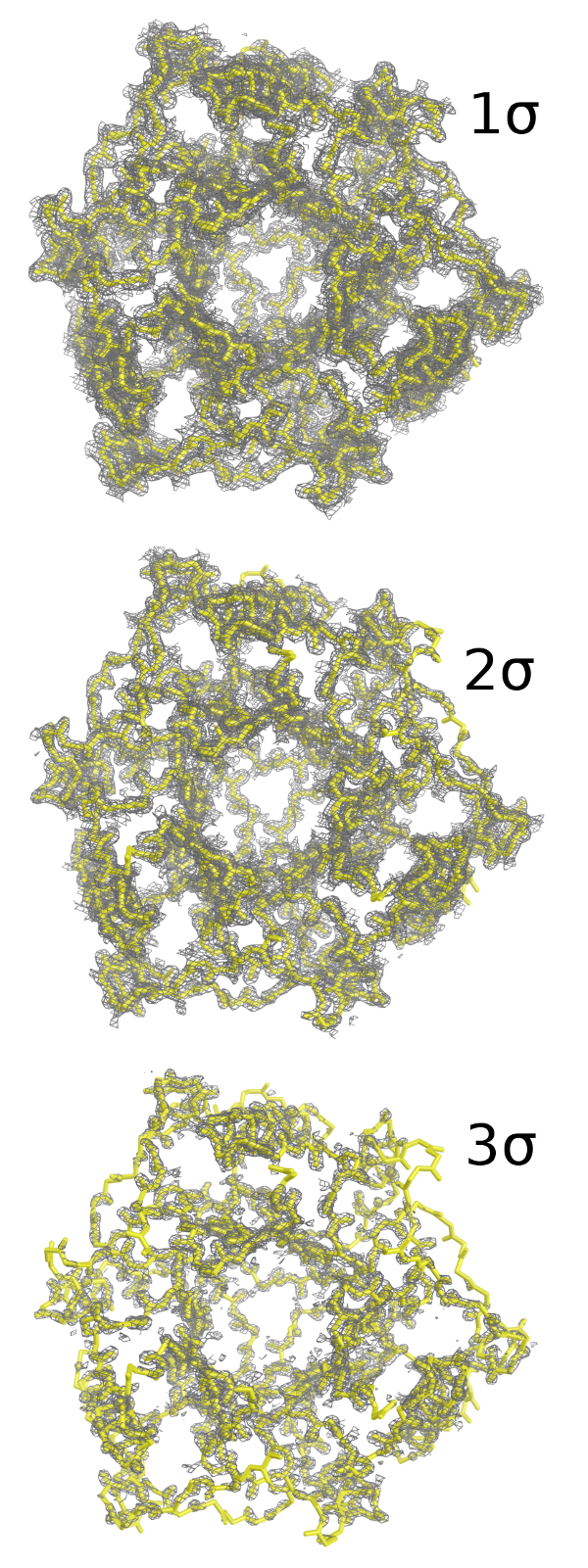
Рис. 1
Затем были построены карты электронной плотности для разных аминокислотных остатков. Первый пример — последовательность His-Lys-Pro, сочетающая в себе положительно заряженные гистидин и лизин с длинным боковым радикалом и пролин. Кольца гистидина и пролина видны на карте электронной плотности очень хорошо, а "хвост" лизина (δC и N атомы) плохо различим дальше при увеличении уровня показанной плотности до 0.7 σ.
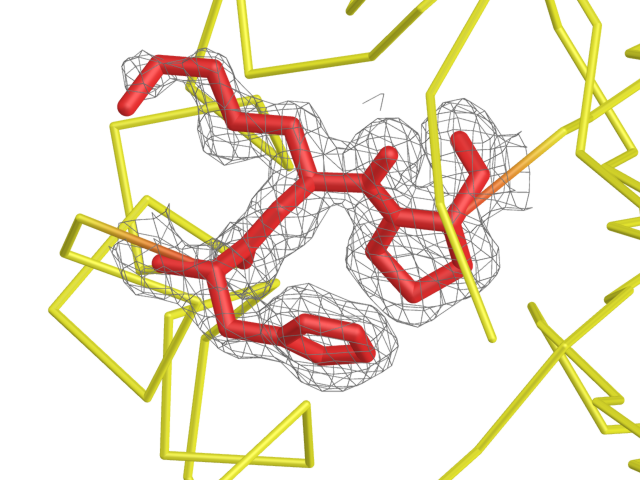
Рис. 2
Другой пример — последовательность Ser-Phe-Val, в котором есть и полярные, и гидрофобные, и даже ароматическая аминокислота. Здесь электронная плотность полностью совпадает с pdb структурой для всех трех аминокислот, отчетливо видел атом кислорода в серине.
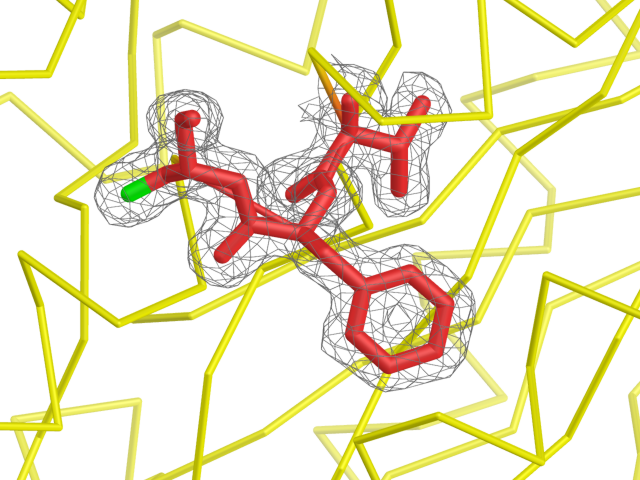
Рис. 3
Наконец, я решила посмотреть, как будет выглядеть на карте электронной плотности атом серы.
На первом же попавшемся остатке цистеина оказалось два атома серы (показаны голубым на картинке ниже). Проверка pdb файла показала, что в структуре действительно прописано два SG атома:
ATOM 1749 CA CYS B 104 7.029 18.689 32.069 1.00 14.11 C ATOM 1750 C CYS B 104 8.350 19.352 32.355 1.00 13.15 C ATOM 1751 O CYS B 104 9.403 18.793 32.048 1.00 13.52 O ATOM 1752 CB CYS B 104 6.868 17.533 33.036 1.00 18.20 C ATOM 1753 SG ACYS B 104 6.920 18.012 34.803 0.75 17.82 S ATOM 1754 SG BCYS B 104 5.569 16.384 32.556 0.25 36.54 S
При этом в цепи А, аналогичной по последовательности, этот цистеин совершенно нормальный. А в такой же цепи С - снова с двумя серами. Как видно, одному из атомов соответствует нормальный радиус электронной плотности. Вокруг второго же ее не видно четко, причем на разных "изоповерхностях" электронной плотности. С другой стороны, что-то там точно есть, и на это нельзя не обращать нимание. Рядом с этим атомом нет никакого другого, чья плотность могла бы слиться с серином и дать такой эффект. UPD: Как выяснилось, такое явление может быть объяснено тем, что в итоговую структуру попадает результат объединения данных из многих ячеек, в 75% которых встречается конформация цистеина, повернутого "вправо" (ACYS), а в остальных 25% — "влево" (BCYS).
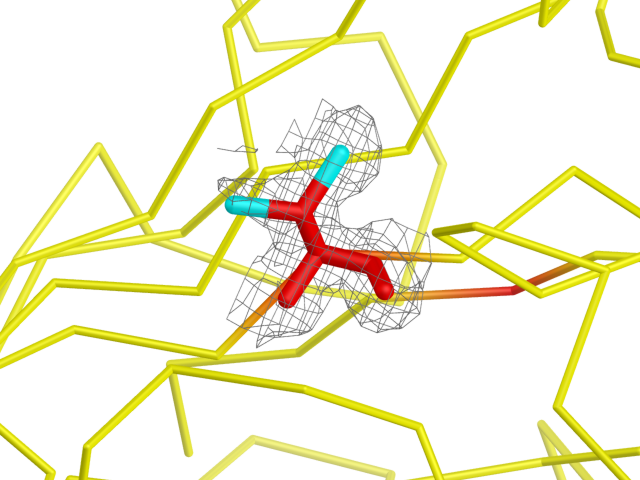
Рис. 4
Вывод о качестве структуры пока что сделать трудно. На ней плохо различимы длинные боковые радикалы, как у лизина. Кроме того, боковые остатки некоторых аминокислот (например, цистеина, описанного выше) имеют разную конформацию в разных ячейках. Пока что сказать что-то конкретное о качестве этой структуры трудно.