
|
|
|
||||||||||||||||||||||||||||||||||||||
Часть 1. Подготовка чтений. Задачей данного практикума является поиск и описание полиморфизмов у человека. Для начала необходимо подготовить полученные с секвенатора чтения для дальнейшей обработки - анализировать их качество и удалить "плохие" участки. С этой целью использовались программы FastQC(анализ качества чтений) и Trimmomatic(очистка чтений). Я использовала команды fastqc chr21.fastq - получила zip архив с отчетом о качестве чтений java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr21.fastq trim_chr21.fastq TRAILING:20 - удалила нуклеотиды с качеством меньше 20 с конца каждого чтения. java -jar /usr/share/java/trimmomatic.jar SE -phred33 trim_chr21.fastq trim2_chr21.fastq MINLEN:50 - выбрала только чтения длиной более 50 нуклеотидов. fastqc trim2_chr21.fastq - провела контроль качества обработанных чтений. Ниже приведены картинки из выдачи программы FastQC, позволяющие сравнить качество чтений до и после обработки. 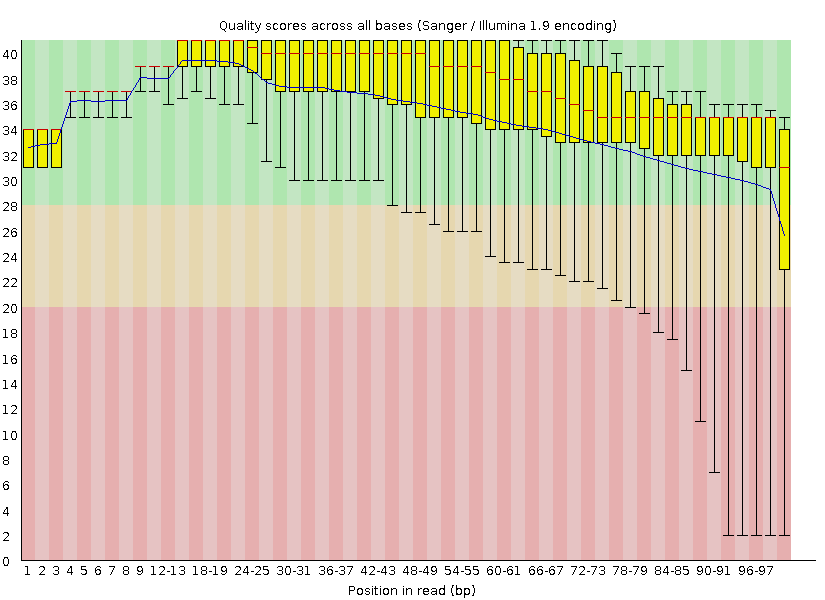
Рис.1 Иллюстрация качества чтений до обработки 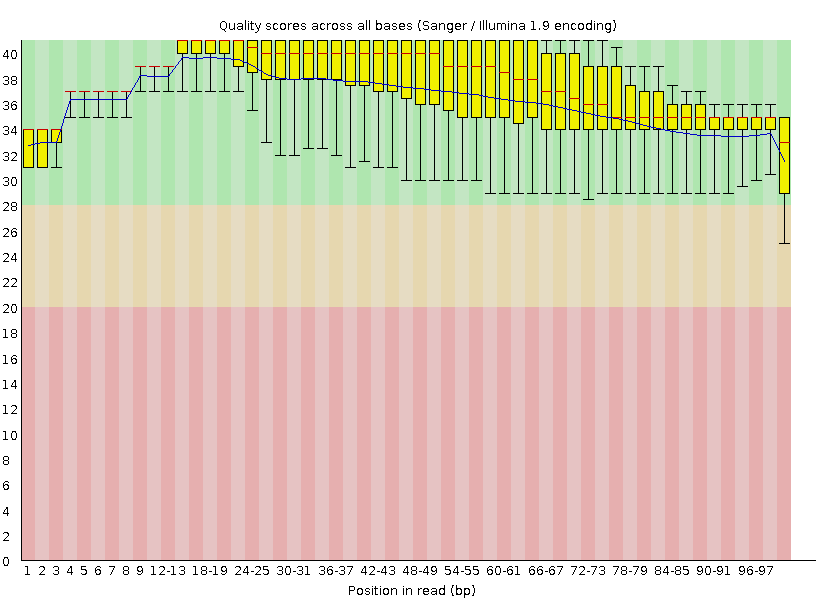
Рис.2 Иллюстация качества чтений после обработки До чистки файл содержал 8158 чтений, после чистки их стало 7858. Получается, что удалилось 300 чтений. В соответствии с примененными мною командами, все эти 300 чтений были длиной менее 50 нуклеотидов. Часть из них могла быть изначально короткой, часть стала короче после первой команды - удаления нуклеотидов с плохим качеством. Часть 2. Картирование чтений. После очистки чтений можно их картировать. Использовалась программа BWA: bwa index chr21.fasta - референсная последовательность была проиндексирована bwa mem chr21.fasta trim2_chr21.fastq > bwa_chr21.sam - построено выравнивание прочтений и референса Полученное выравнивание я проанализировала программой samtools: samtools view -b bwa_chr21.sam -o sam_outf.bam - перевела файл в .sam формате в формат .bam(необходимо для дальнейшей работы) samtools sort -o sam_sort1.bam -T out.prefix sam_outf.bam - отсортировала выравнивание чтений с референсом по координате в референсе начала чтения samtools index sam_sort1.bam - проиндексировала отсортированный файл samtools idxstats sam_sort1.bam - посмотрела, сколько чтений откартировалось на геном. Выдача samtools idxstats показала, что все 7858 чтений картировались. Часть 3. Анализ SNP. С помощью samtools mpileup я получила файл с полиморфизмами в формате .vsf, далее с помощью команды bcftools call -cv chr21snp.bcf -o chr21snp.vcf перевела его в формат .vcf. Из полученного файла - chr21snp.vсf - я взяла полиморфизмы для описания. Всего нашлось 82 полиморфизма и 6 инделей. Для более наглядного представления данных я перевела таблицу из текстового файла в excel - ссылка на html-вид таблицы. Голубым выделены описываемые полиморфизмы. 1) Замена С(в рефренсе) на Т(в чтениях) в 16334658 позиции. Качество чтений в данном месте - 166.009(хорошее), глубина покрытия данного места - 16(средняя). 2) Замена: в референсе был нуклеотид Т, в чтениях G. Позиция - 16334963. Качество чтений в данном месте - 225.009(очень хорошее). Глубина покрытия - 75(очень хорошая). 3) Опять замена нуклеотида С(в референсе) на Т(в чтениях). Позиция - 16335402. Качество чтений в данном месте - 16,1143(достаточно низкое). Глубина покрытия - 12(средняя). Аннотация snp.Получив список snp, их можно аннотировать. Для этого я использовала программу annovar. Сначала было необходимо получить файл в формате, с которым умеет работать эта программа: perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr21snp.vcf -outfile chr21snp.avinput После этого я аннотировала имеющиеся у меня SNP c использованием 5 баз данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar(команды приведены в таблице 1). Refgene После использования команды было получено три файла. Один из них, с "форматом" .variant_function содержит информацию обо всех SNP, в нем они подразделяются на группы в зависимости от того, где произошла замена - в экзоне, интроне или 5' 3' некодирующих областях. Другой, с "форматом" .exonic_variant_function содержит информацию только о нуклеотидах, попавших в экзоны. У меня 4 SNP попали в экзоны, 69 в интроны и 9 в некодирующие концевые области. О SNP, попавших в экзоны.Один из полиморфизмов оказался в гене белка, взаимодействующего с рецепторами ядра у человека(Homo sapiens nuclear receptor interacting protein 1). Три других попали в ген белка А, ассоциированного с убиквитином и содержащего SH3 домен(Homo sapiens ubiquitin associated and SH3 domain containing A (UBASH3A)). Из четырех полиморфизмов, попавших в экзоны, замену аминокислот вызвала только замена А на G в 43824106 позиции. Замены, попавшие в индели и концевые некодирующие участки, отнесены к тем же генам и к гену белка ацетилтрансферазы(AGPAT3 1-acylglycerol-3-phosphate O-acyltransferase 3). Интересно отметить, что в экзонах полиморфизмов значительно меньше, чем в некодирующих областях. Этому имеется понятное биологическое объяснение - ошибки в кодирующих последовательностях с большой вероятностью могут повлиять на жизнедеятельность организма, а ошибки в "молчащем" генетическом материале никак не отражаются на белках. dbsnp После команды по этой базе данных было опять получено три файла. В файле с форматом dropped содержатся snp, имеющие rs(идентификатор замены в dbsnp), в файле filtered - не имеющие. rs имеют 60 SNP, не имеют 22. 1000 genomes В файле dropped были SNP, аннотированные в 1000 genomes, для них была указана частота встречаемости. Она находится в пределах от 0,01 до 0,8(для одного нуклеотида оказалась 1). Таких полиморфизмов оказалось 59. Не аннотированными в данной БД оказались 23. Базы, предоставляющие клиническое описание SNP(GWAS, Clinvar) В базе данных GWAS аннотировано 3 полиморфизма из моего списка. Один из них связан с когнитивными функциями(cognitive performance), другой с диабетом первого типа, третий с уровнем фосфолипидов в плазме крови. В базе данных Clinvar аннотированных полиморфизмов не нашлось. Итог: Я сделала общую таблицу по snp с характеристиками, полученными из различных баз данных(Clinvar не представлена, так как в ней никаких snp из моих файлов не оказалось) - таблица. |
|||||||||||||||||||||||||||||||||||||||
© Шугаева Т.Е. 2015