Парное выравнивание белковых последовательностей и применение алгоритмов парных выравниваний к цитратсинтазе I
Модель эволюции белковых последовательностей
Задача и методы: В данном задании вручную произведено парное выравнивание последовательности моего белка с несколькими 20-ти аминокислотными участками, которые были предварительно получены как результат работы скрипта evolve_protein.pl при использовании различных параметров. Скрипт иллюстрирует появление случайных мутаций в последовательности белка из-за ошибок в деятельности ДНК-полимеразы и использует для своей работы несколько параметров: (i) вероятность возникновения мутации в данном месте аминокислотной последовательности {параметр -i}; (ii) вероятность того, что при наличии мутации произойдёт замена, а не делеция или вставка другой аминокислоты {параметр -r}.Для работы скрипта также определены следующие дополнительные параметры:
- -f: создаёт полноразмерные последовательности
- -t: задает количество создаваемых последовательностей
- -g: задает количество "поколений мутагенеза", то есть проходов по исходной последовательности
- >0|simulation_result|change=0.6|replace=0.6|generations=1 SVLSVQFIALRFNLTYFPKT
- >0|simulation_result|change=0.6|replace=0.8|generations=1 PTFAESLQDFMDRDAGRNCS
- >0|simulation_result|change=0.4|replace=0.8|generations=1 MSDIGEEHNFEAYKEKLTGE
- c более или менее неполярным боковым радикалом (синий цвет)
- c положительно заряженным радикалом (светло-зелёный цвет)
- c отрицательно заряженным радикалом (красный цвет)
- c незаряженным полярным радикалом (жёлтый цвет)
- c ароматическим боковым радикалом (голубой цвет)
| Выравнивание | Identity | Similarity | Alignment weight |
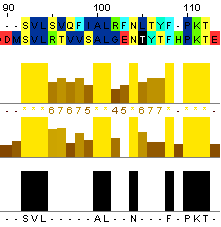 | 10/21*100% = 47,6% | 10/21*100% = 47,6% | 14 |
 | 9/21*100% = 42,86% | 13/21*100% = 61,9% | 17 |
 | 13/22*100% = 59% | 14/22*100% = 63,64% | 44 |
Обсуждение результатов:
- Наблюдается незначительное различие между similarity и identity, что может быть обусловлено тем, что рассмотрено сравнительно большое количество групп аминокислот, что привело к уменьшению similarity. Вероятно, similarity является наименее объективным параметром сравнения выравнивания из трёх предложенных, так как зависит от выбора групп аминокислот, считающихся схожими в конкретном случае. Но стоит сказать, что использование similarity весьма удобно при сравнении функциональных свойств двух белков, что правда имеет к нашей модели эволюции белковых последовательностей косвенное отношение.
- Выравнивания 1 и 2 более или менее сходны по всем трём параметрам оценки, что обусловлено одинаковой вероятностью замены в 60%. Как и предполагалось, в выравнивании 3 из-за того, что вероятность внесения каких-либо изменений составляла всего 40%, больше половины аминокислот остались консенсусными для двух последовательностей. Это даёт последнему выравниванию наибольшие результаты по всем трём критериям оценки качества выравнивания.
Выравнивание цитратсинтазы I с её ортологами
В данном разделе приведены результаты, полученные при проведении выравнивания моего белка с его ортологами (об ортологах см. соответствующий раздел второго семестра). Сначала был составлен файл, содержащий последовательности трёх белков в FASTA-формате.Затем с использованием программы MUSCLE было проведено выравнивание последовательностей. Для каждой пары выровненных последовательностей были определены характеристики с использованием программы infoalign из пакета EMBOSS.
Описание программы infoalign:
Программа выводит данные о множественном (в том числе и о парном) выравнивании последовательностей. Важнейшие опции:
- -gaps показывает количество пропусков
- -gapcount показывает число аминокислот, соотнесённых с пропуском
- -idcount показывает количество идентичных позиций
- -simcount показывает число похожих позиций
- -difcount показывает число различных позиций
| Name | Sequence Length | Aligned Length | Gaps | Gap Length | Identity | Similarity | Difference | % Change | Weight |
|---|---|---|---|---|---|---|---|---|---|
| CISY_BACSU/1-366 | 366 | 377 | 3 | 11 | 366 | 0 | 0 | 2.917772 | 1.000000 |
| CISY_BACCO/1-373 | 373 | 377 | 1 | 4 | 142 | 73 | 158 | 62.334217 | 1.000000 |
| Name | Sequence Length | Aligned Length | Gaps | Gap Length | Identity | Similarity | Difference | % Change | Weight |
|---|---|---|---|---|---|---|---|---|---|
| CISY_BACSU/1-366 | 366 | 377 | 3 | 11 | 366 | 0 | 0 | 2.917772 | 1.000000 |
| Q81QS0_BACAN/1-372 | 372 | 378 | 2 | 6 | 146 | 72 | 154 | 61.375660 | 1.000000 |
| Name | Sequence Length | Aligned Length | Gaps | Gap Length | Identity | Similarity | Difference | % Change | Weight |
|---|---|---|---|---|---|---|---|---|---|
| CISY_BACCO/1-373 | 373 | 377 | 1 | 4 | 373 | 0 | 0 | 1.061008 | 1.000000 |
| Q81QS0_BACAN/1-372 | 372 | 378 | 2 | 6 | 271 | 35 | 66 | 28.306879 | 1.000000 |
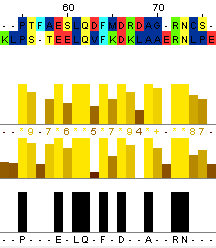
Выравнивание трёх последовательностей было окрашено по схеме Clustalx с использованием порога идентичности в 66% (позволяет окрашивать только те позиции, где идентичными являются 2 или 3 аминокислоты во множественном выравнивании (см. рис. 1).
 |
| Рис. 1 Участок выравнивания с 1-ой по 122-ю позицию выравнивания, раскрашенный по схеме Clustalx с порогом идентичности в 66%. |
Дата последнего обновления: 31.03.2013
© Dmitry Travin, 2012