Алгоритмы выравнивания в применении к белку CISY_BACSU
Сравнение матриц аминокислотных замен
- Что такое матрицы аминокислотных замен? Для того, чтобы оценить насколько хорошо то или иное выравнивание нескольких последовательностей прибегают к помощи матриц аминокислотных замен - таблиц, в которых для каждой пары аминокислот указано число, которое указывает на то насколько вероятна замена одной аминокислоты на другую. Если замена вероятна, то число положительно, если же нет, то перед ним стоит знак "-". Матрица строится на основании встречаемости определённой аминокислоты в базе данных аминокислотных последовательностей (для BLOSUM62 - база данных BLOCKS).
- Что обозначают числа после названия матрицы? В процессе построения матрицы замен необходимо учесть вклад последовательностей, которые являются близкими или даже одинаковыми, чтобы их влияние не сдвигало вероятности. На помощь приходит кластеризация - приём объединения последовательностей с определённым уровнем идентичности (этот порог в % и указан после названия) в единый кластер. Дальнейшая работа проводится со всем кластером как с единым целым.
- Почему существуют разные матрицы? Использование для различных групп белков различных матриц обусловлено различиями во встречаемости в них аминокислот. Это в свою очередь в большой степени обусловлено тем, где локализован в клетке определённый белок. Например, для мембранных белков, в которых есть трансмембранные тяжи из гидрофобных остатков и некоторые другие особенности, используются матрицы типа PHAT, в то время как матрицы типа BLOSUM применимы в основном к цитоплазматическим белкам(по данным о которых и составлена изначально база данных BLOCKS). матрица PHAT
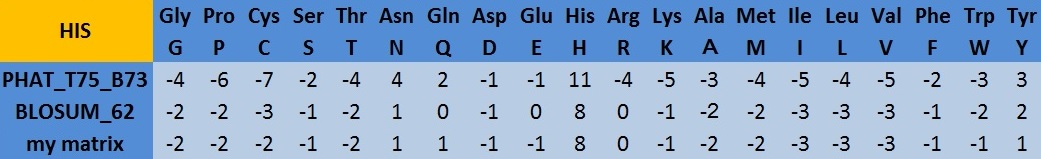 |
| Таблица 1 Сравнение значений аминокислотных замен гистидина для трёх различных матриц. |
Анализ полученных данных сравнения
- Как отличаются величины замены аминокислоты на саму себя? Почему? Во всех трёх случаях это значение (11,8,8 - см. таблицу 1) представляет сравнительно большое положительное число, что объясняется, так как замена аминокислоты саму на себе это по сути неизменность последовательности, что, разумеется, встречается в эволюции белковых крайне часто.
- Как отличаются величины замены аминокислоты на близкие по химическим свойствам? Почему? По химическим свойствам к гистидину наиболее близки лизин и аргинин, так как они тоже обладают положительно заряженным боковым радикалом. Но несмотря на близость химических свойств, частотные данные говорят о том, что у цитоплазматисческих белков вероятность равна случайной (значения 0 для Arg в BLOSUM и my) или даже меньше случайной (-1 для Lys). В это же время в PHAT эти значения отрицательны, что показывает низкую частоту подобных замен.
- Как отличаются величины замены аминокислоты на аминокислоты из других функциональных групп? Почему? Для таких замен характерны наиболее отрицательные значения во всех матрицах. В основном это неполярные остатки (Gly, Pro, Cys, Val, Ile, Leu).
- Интересные замечания Для матрицы PHAT по всем значениям характерны более крупные значения (по модулю). Зная о том, как рассчитывается такая матрица, можно заключить, что отношение реальной вероятности встречи замены к случайной для мембранных белков выше чем для цитоплазматических. Это может быть связано с тем, что последовательности таких белков имеют в среднем больше консервативных участков по сравнению с белками, локализованными в цитозоле.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Программа needle, представляющая из себя часть пакета EMBOSS, является инструментом для выравнивания двух последовательностей с помощью алгоритма глобального выравнивания Нидлмана-Вунша <статья>. Программа использует для проведения выравнивания следующие параметры:- -gapopen штраф за открытие пропуска [по умолчанию равен 10]
- -gapextend штраф за удлинение пропуска на одну позицию [по умолчанию равен 0.5]
- -datafile используемая матрица [по умолчанию BLOSUM62 для белковых и EDNAFULL для нуклеотидных последовательностей]
- -endopen штраф за открытие конечного пропуска [по умолчанию равен 10]
- -endextend штраф за удлинение конечного пропуска на одну позицию [по умолчанию равен 0.5]
Основное отличие между алгоритмами локального и глобального выравнивания состоит в том, что в первом случае нет штрафа за открытие начального пропуска. Это позволяет выравнивать последовательности не прямо, а подбирать наилучшее совпадение участка.
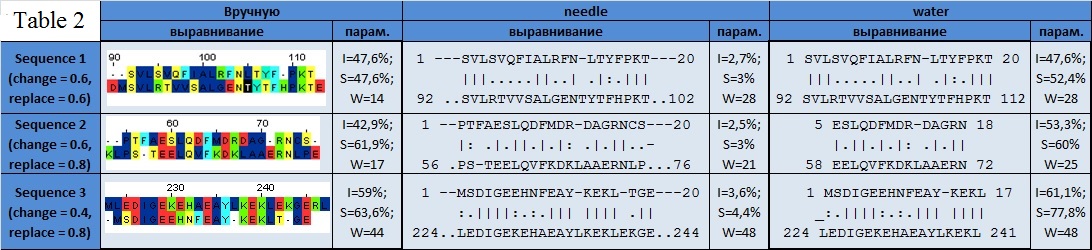 |
| Таблица 2. Результаты выравнивания вручную и с использованием программ needle и water при стандартных значениях опций (для выравниваний также указаны параметры: S=similarity, I=identity, W=alignment weight). |
Описание полученных различными методами выравниваний:
- Первое, что необходимо отметить, это различная длина выравниваний полученных разными методами. Вручную и с помощью программы needle весь участок из 20-ти аминокислот был выровнен с изначальным белком, в то время как программа water во втором и третьем случае, выполняя задачу локального выравнивания, просто удалила плохо выравниваемый участок последовательности из выравнивания. Этого можно избежать, понижая штраф за внесение и продолжение пропусков.
- Второе важное наблюдение состоит в том, что при проведении глобального выравнивания большого белка и короткого фрагмента такие показатели как identity и similarity считаются исходя не из длины выровненной части, а из длины большей последовательности. Поэтому мы видим результаты, которые не могут характеризовать сам выровненный участок.
- Сравнение параметров, характеризующих качество выравнивания, показывает похожесть identity и similarity между выравниваниями вручную и с использованием water. Оценочный балл, рассчитанный исходя из матрицы для различных выравниваний сильно расходится, что связано с использованием различных баллов за открытие и продолжение пропуска в различных случаях.
Дата последнего обновления: 03.04.2013
© Dmitry Travin, 2012