Комплексы ДНК с белком
Использование команды define
Начнём с использования команды define для выделения различных наборов (sets) атомов. В качестве упражнения напишем скрипт который последовательно (переход между вариантами осуществляется нажатием клавиши Enter) позволит нам выделить следущие части в структуре:- весь комплекс нуклеиновой кислоты и белка
- только нуклеиновая кислота в проволочной модели
- атомы кислорода 2'-дезоксирибозы (set1)
- атомы кислорода в остатке фосфорной кислоты (set2)
- атомы азота в азотистых основаниях (set3)
Поиск ДНК-белковых контактов в заданной структуре
Интересно проанализировать по каким группам и вообще как происходит контакт белка и нуклеиновой кислоты из нашей структуры pdb. Для этого выделим все те места, где расстояние между различными функциональными группами из белка и ДНК составляет:- не более 3,5 ангстрем между условно "полярными" атомами (азот, кислород)
- не более 4,5 ангстрем между условно "неполярными" атомами (углерод, фосфор, сера)
- атомы ДНК, участвующие в полярном взаимодействии - синий цвет
- атомы ДНК, участвующие в неполярном взаимодействии - желтый цвет
- атомы белка, участвующие в полярном взаимодействии - красный цвет
- атомы белка, участвующие в неполярном взаимодействии - зелёный цвет
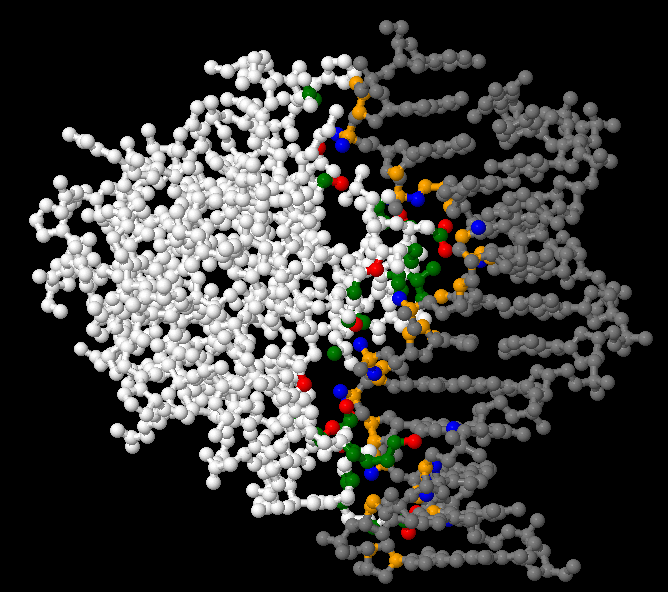 |
Рис. 1 Выделение атомов, гипотетически участвующих во взаимодействиях нуклеиновой кислоты (ДНК) и белка с использованием команд скрипта 2. Днк показана серым, белок белым, а выделение а выделение взаимодействующих атомов показано в соответствии со схемой в тексте. |
Важно отметить, что такое выделение атомов не достаточно точно отражает реальную ситуацию, так как в действительности далеко не все пары атомов, расстояние между которыми 3,5 или же 4,5 ангстрем (для полярного и неполярного взаимодействий соответственно), будут образовывать некое подобие связи. Это может происходить в силу многих причин: наличием между ними других атомов, "неудобным расположением соседних атомов, мешающих контакту и т.п. Таким образом в таблице приведены подсчёты максимальных значений для числа контактов каждого типа.
| Контакты атомов белка с: | Полярные | Неполярные | Всего |
| 1) остатками 2'-дезоксирибозы | 1 | 12 | 13 |
| 2) остатками фосфорной кислоты | 9 | 6 | 15 |
| 3) остатками азотистых оснований со стороны большой бороздки | 1 | 8 | 9 |
| 4) остатками азотистых оснований со стороны малой бороздки | 2 | 0 | 2 |
Проанализируем данные, представленные в таблице. Во-первых, считаю почти невозможным образование неполярных взаимодействий с участием остатка фосфорной кислоты, так как полярные кислороды будут явно тому препятствовать. Во-вторых, отмечу, что из таблицы явно видно преимущественное участие в образовании взаимодействий именно атомов остова, по сравнению с основаниями в центре двойной спирали. В-третьих, атомы оснований, обращённые в большую бороздку, образуют больше связей.
Использование программы nuclpot
Для получения наглядной схемы контактов в молекуле ДНК была использована программа nuclpot. Она выдаёт несколько файлов различного формата. Но нас в первую очередь интересует популярная схема, отражающая контакты каждого нуклеотида из ДНК с различными молекулами окружения (вода, белок, ионы). Визуализация выполнена с помощью программы GSview, которая открывает файлы формата .ps. 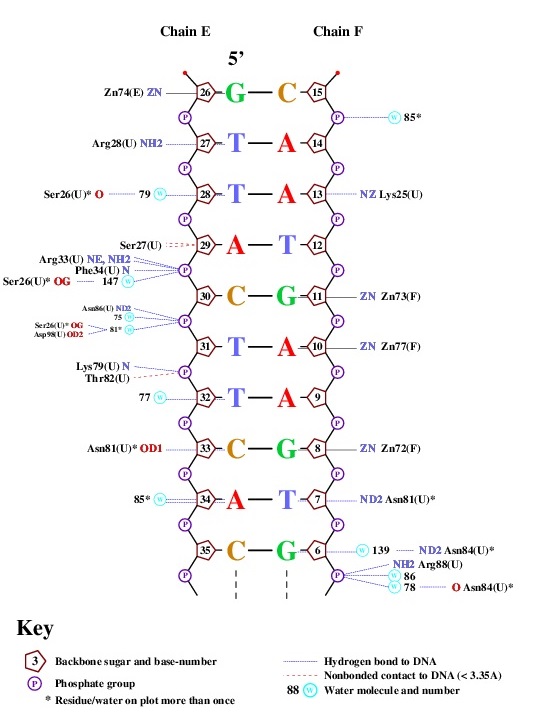 |
Рис. 2 Иллюстрация контактов цепей ДНК, полученная с помощью программы nuclpot |
 |
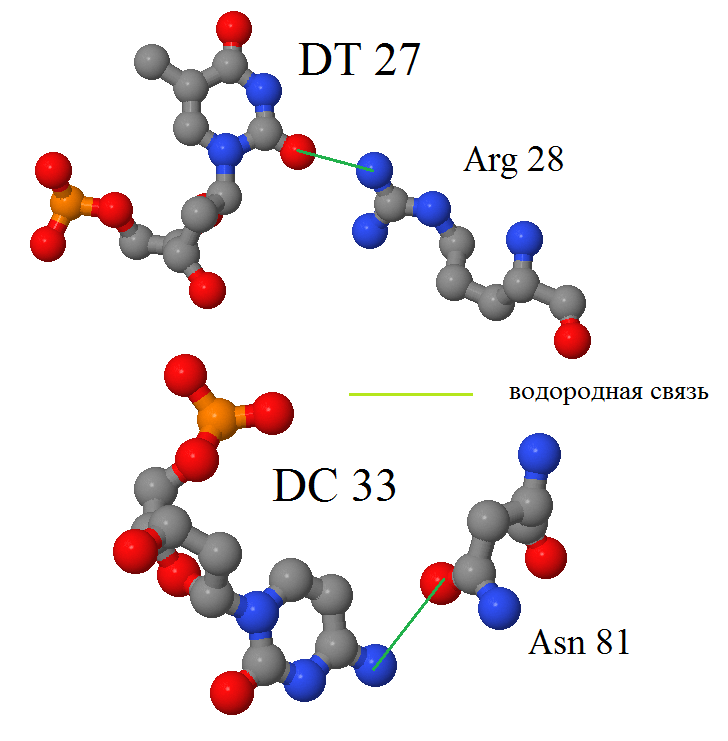 |
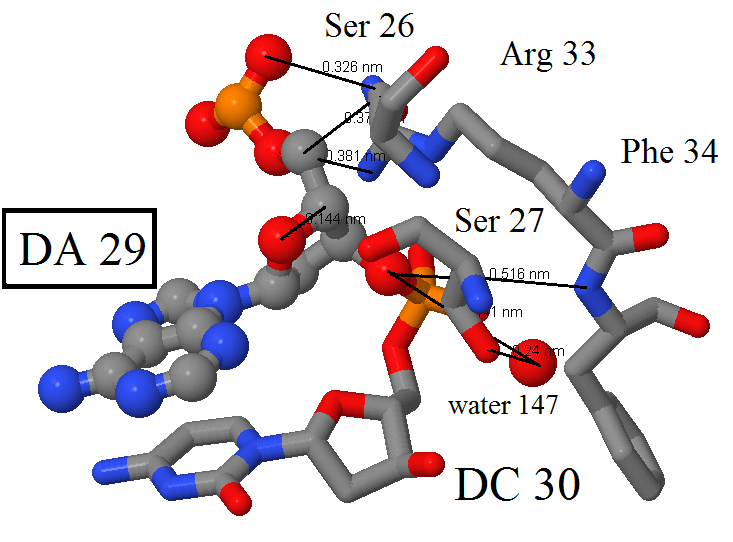
Рис. 3 Взаимодействие DA 29 с белком. Показаны 4 аминокислотных остатка и молекула воды, стабилизированная в структуре. Чёрным показаны различные типы взаимодействий: как водородные связи, так и неполярные виды связи. | Рис. 4 Примеры взаимодействий (DT 27 и DC 33), когда в образовании водородных связей участвует остаток азотистого основания (обеспечение специфичности). |
Предсказание вторичной структуры транспортной РНК
В то время как для ДНК вторичная структура в большинстве случаем вполне ясна, так как она получается при комплиментарном объединении двух цепочек нуклеотидов, то для РНК она может быть очень и очень сложной, содержащей в своём составе комбинации многих разнообразных элементов. Именно этому будет посвящена вторая часть этой странички.Предсказание вторичной структуры методом поиска инвертированных повторов
В этом задании мы используем новые подходы к выявлению участков спаривания во вторичной структуре тРНК:1) команду einverted, которая позволяет найти инвертированные участки в последовательности:
работает эта команда с файдом последовательности в формате .fasta, кроме того требуется задать некоторые начальные параметры, а именно:
- Штраф за пропуск (Gap penalty): -5
- Минимальное количество набранных очков (Minimum score threshold): 20
- Штраф за несоответствие (Mismatch penalty): -10
- Количество очков за правильное соответствие (Match score): 10
При работе с командой mfold обратим внимание на некоторый параметр P, который показывает, на сколько процентов по энергии может отличаться предсказанная структура от оптимальной, таким образом вполне очевидно, что с ростом параметра Р будет происходить увеличение количества возможных вариантов строения макромолекулы.
При росте параметра P увеличивалось количество различных предсказаний, но те, что действительно похожи на реальную структуру транспортной РНК, появлялись преимущественно при низких значениях P (0-5%). Один из таких результатов представлен ниже.
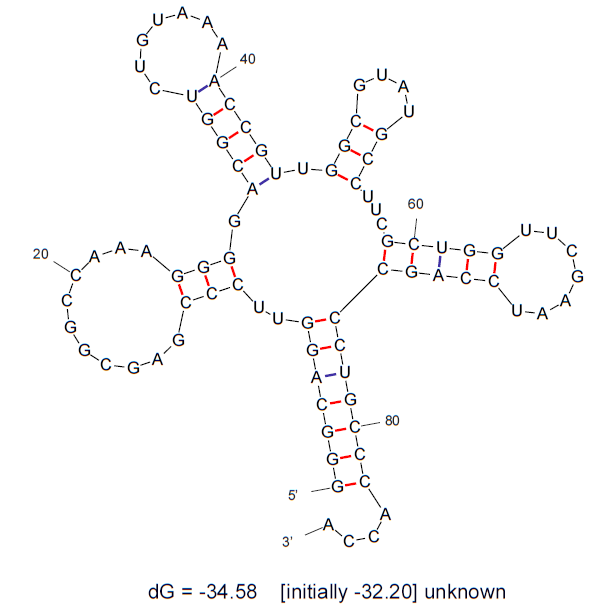 |
Рис. 4 Результат предсказания вторичной структуры транспортной РНК с использованием алгоритма Зукера. Использовался веб интерфейс для запуска программы mfold Mobyle @Pasteur |
| Участок структуры / Способ определения | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | |||
| D-стебель | |||
| T-стебель | |||
| Антикодоновый стебель | |||
| Общее число канонических пар нуклеотидов |
Дата последнего обновления: 01.10.2013
© Dmitry Travin, 2013