Standalone BLAST
Поиск в геноме участков, кодирующих белки, похожие на заданный
В этом задании попытаемся найти гомолог цитратсинтазы из сенной палочки в геноме Listeria monocytogenes, не используя при этом аннотацию генома. Будем задействовать алгоритм tblastn: поиск известной белковой последовательности в формальной трансляции нуклеотидного банка данных.- Сначала создадим индексные файлы пакета BLAST+ командой makeblastdb -in lm_genome.fasta -dbtype nucl -out lm.fasta, получаем триф файла с расширениями .nhr, .nin и .nsq.
- Теперь проведём blast нашей известной последовательности query.fasta против созданной базы командой tblastn -query query.fasta -db lm.fasta -out result.txt -evalue 0.001 -outfmt 6.
- Получили один результат работы blast со следующими характеристиками:
Таблица 1. Поиск гомологов белка CISY_BACSU в геноме Listeria monocytogenes. Характеристики полученных при поиске алгоритмом tblastn результатов.
Число находок с E-value < 0,001 1 E-value лучшей находки 2E-69 Название последовательности с лучшей находкой AL591979 (7 из 12 фрагментов, на которые разбит геном бактерии) Координаты лучшей находки 197001-195937 Доля последовательности белка, вошедшая в выравнивание с лучшей находкой 100% (длина выравнивания 371, а не 366 в силу наличия нескольких пропусков)
Поиск гомологов некодирующих последовательностей программой BLASTN
В этом задании мне предстоит, имея файл с последовательностями всех тРНК из генома сенной палочки, узнать сколько гомологов находится в геноме Listeria monocytogenes для этих последовательностей.- Запустим поиск алгоритмом blastn по базе, содержащей геном бактерии, используя в качестве запроса файл с последовательностями тРНК из сенной палочки: blastn -query trna_bacsu.fasta -db lm.fasta -evalue 0.01 -task blastn -outfmt 7. Просмотрим содержание полученного файла: ССЫЛКА.
- Теперь попробуем с помощью команд bash посчитать количество результатов по каждой из тРНК. Сначала получаем колонку-список всех последовательностей в запросе: grep ">" trna_bacsu.fasta. Затем составим скрипт для подсчёта числа результатов: ССЫЛКА. С помощью FAR переведём его в формат для Unix и активируем командой chmod +x. Запуск скрипта через Putty производим так: ./script.sh, если находимся в той директории, где находится и файл скрипта.
- Результаты выполнения задания сохраняем в файл Exсel, в виде таблицы с двумя столбцами: название исходной записи для тРНК из B. subtilis и количество соответствующих ей гомологов из генома L.monocytogenes. ССЫЛКА
Поиск гомологов при изменённых параметрах программы BLASTN
Теперь запустим программу blastn ещё дважды, но на этот раз с изменёнными параметрами:1) -reward 5 (бонус за одинаковый нуклеотид в выравнивании),-penalty -4 (штраф за mismatch), -gapopen 10 (штраф за открытие пропуска) -gapextend 6 (штраф за продолжение пропуска)
blastn -query trna_bacsu.fasta -db lm.fasta -evalue 0.01 -task blastn -outfmt 72) те же параметры, что и до этого, но с использованием минимальной длины "слова" для поиска (4 нуклеотида), -word_size 4
-reward 5 -penalty -4 -gapopen 10 -gapextend 6 -out final2.txt
blastn -query trna_bacsu.fasta -db lm.fasta -evalue 0.01 -task blastn -outfmt 73) поиск со стандартными параметрами штрафов и бонусов, но минимальной длиной слова в 4 нуклеотида
-reward 5 -penalty -4 -gapopen 10 -gapextend 6 -word_size 4 -out final3.txt
blastn -query trna_bacsu.fasta -db lm.fasta -evalue 0.01 -task blastn -outfmt 7Теперь ко всем трём полученным файлам применим написанный во втором задании скрипт и добавим новые результаты в таблицу Excel ССЫЛКА.
-word_size 4 -out final4.txt
Анализ полученных результатов
Итак, при внесении изменений (как в штрафы, так и в длину слова), наблюдается увеличение суммарного числа найденных гомологов (Рис. 1). Особенно оно заметно при внесении в исходный запрос обоих описанных изменений.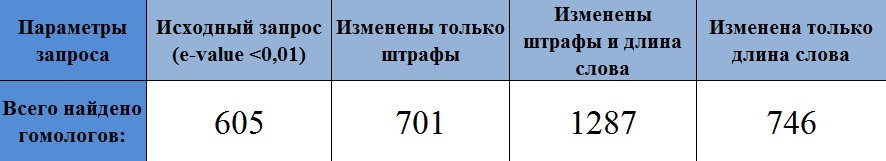 |
| Рис. 1 Зависимость количества находок команды BLAST от исходных параметров запроса. |
Теперь выберем одну из записей для тРНК B. subtilis (BSn5_t20982 tRNA-Thr) и находку blast (embl|AL591974 43053-43108), которая получается при запросах №№ 1 и 2 (где изменены параметры для штрафов) и не находится при стандартных параметрах матрицы (исходный запрос и запрос №3). Вырежем гомологичный кусочек и командой seqret, проведём выравнивание с помощью needle.
Результаты:
1) Выравнивание и его характеристики, приводимые в выдаче blast:
Score = 33.0 bits (124), Expect = 0.005 Identities = 40/56 (71%), Gaps = 1/56 (2%) Query 8 TAGCTCAGCA-GGTAGAGCACTTCCATGGTAAGGAAGAGGTCAGCGGTTCGAGCCC 62 ||||||||| |||||||||||| | | | ||| |||| ||||||| || Sbjct 43053 TAGCTCAGCTTGGTAGAGCACTTGGTTTGGGACCAAGGGGTCGCAGGTTCGAATCC 431082) Полученное с помощью команды needle выравнивание и его характеристики:
Gap_penalty: 10.0, Extend_penalty: 0.5 Identity: 42/68 (61.8%) Similarity: 42/68 (61.8%) Gaps: 20/68 (29.4%) Query 1 gcttccatagctcagc-aggtagagcacttccatggtaagg----aagag 45 ||||||||| .||||||||||| ||||..|| |||.| Sbjct 1 -------TAGCTCAGCTTGGTAGAGCACT----TGGTTTGGGACCAAGGG 39 Query 46 gtcagc-ggttcgagc-- 60 ||| || |||||||.. Sbjct 40 GTC-GCAGGTTCGAATCC 56Из полученных данных видно, что выравнивания blast и needle схожи как визуально, так и по большинству параметров. Если же посмотреть аннотацию этого участка в геноме L.monocytogenes, то можно увидеть, что именно в этих границах закодирована тРНК, но не треониновая, как мы искали, а пролиновая. Это может говорить о том, что "при смягчении" параметров поиска (изменение стоимости штрафов и бонусов) в выдаче находятся гомологи - тоже гены тРНК, но при этом не того типа, что в запросе. Именно, на них, вероятно можно списать увеличение числа находок blast в запросах 1,2.
Время работы программы BLAST
Проанализируем время, затрачиваемое программой на тот или иной запрос:Результаты работы команды time:
- Исходный запрос: real 0m 0.590s, user 0m 0.340s
- Запрос №1 (с изменёнными параметрами штрафов): real 0m 0.721s, user 0m 0.448s
- Запрос №2 (с изменёнными параметрами штрафов и длиной слова): real 0m 29.438s, user 0m 28.604s
- Запрос №3 (изменена только длина слова): real 0m 22.013s, user 0m 21.648s
Дата последнего обновления: 14.12.2013
© Dmitry Travin, 2013