Реконструкция филогенетических деревьев
Таксономическое положение выбранных бактерий
C помощью соответствующего сервиса NCBI для семи выбранных в предыдущем практикуме видов бактерий было определено их систематическое положение (см. таблицу 1).Таблица 1. Систематическое положение рассматриваемых бактерий (по NCBI-taxanomy)
| Полное название бактерии | Мнемоника | Систематическое положение |
| Bacillus anthracis | BACAN | Bacteria; Firmicutes; Bacilli; Bacillales; Bacillaceae; Bacillus; Bacillus cereus group |
| Clostridium botulinum | CLOB1 | Bacteria; Firmicutes; Clostridia; Clostridiales; Clostridiaceae; Clostridium |
| Enterococcus faecalis | ENTFA | Bacteria; Firmicutes; Bacilli; Lactobacillales; Enterococcaceae; Enterococcus |
| Geobacillus kaustophilus | GEOKA | Bacteria; Firmicutes; Bacilli; Bacillales; Bacillaceae; Geobacillus |
| Lactobacillus delbrueckii | LACDA | STRPNBacteria; Firmicutes; Bacilli; Lactobacillales; Lactobacillaceae; Lactobacillus |
| Listeria monocytogenes | LISMO | Bacteria; Firmicutes; Bacilli; Bacillales; Listeriaceae; Listeria |
| Streptococcus pneumoniae | STRPN | Bacteria; Firmicutes; Bacilli; Lactobacillales; Streptococcaceae; Streptococcus |
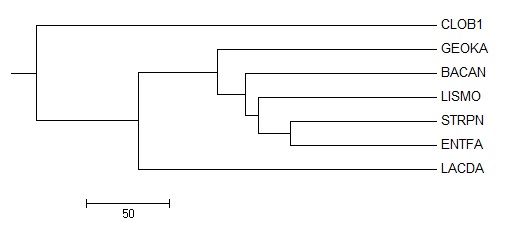
Таким образом, на построенном нами ранее дереве можно произвести сопоставление отдельных ветвей с систематическими группами:
- {BACAN,GEOKA} - Bacillaceae
- {BACAN,GEOKA,LISMO} - Bacillales
- {LACDA,ENTFA,STRPN} - Lactobacillales
Подготовка к построению филогенетического дерева по последовательностям белков
В качестве белка, по которому впоследствии будет строиться филогенетическое дерево, был выбран рибосомальный белок RL3. Из Swiss-prot с помощью команды seqret @list.txt sequence.fasta получаем последовательности белков RL3 по составленному списку их идентификаторов (ССЫЛКА). Получен следующий файл: ССЫЛКА.Далее для полученных последовательностей было получено выравнивание MUSCLE (использовался веб-интерфейс программы http://www.ebi.ac.uk/Tools/msa/muscle) и импортировано в JalView.
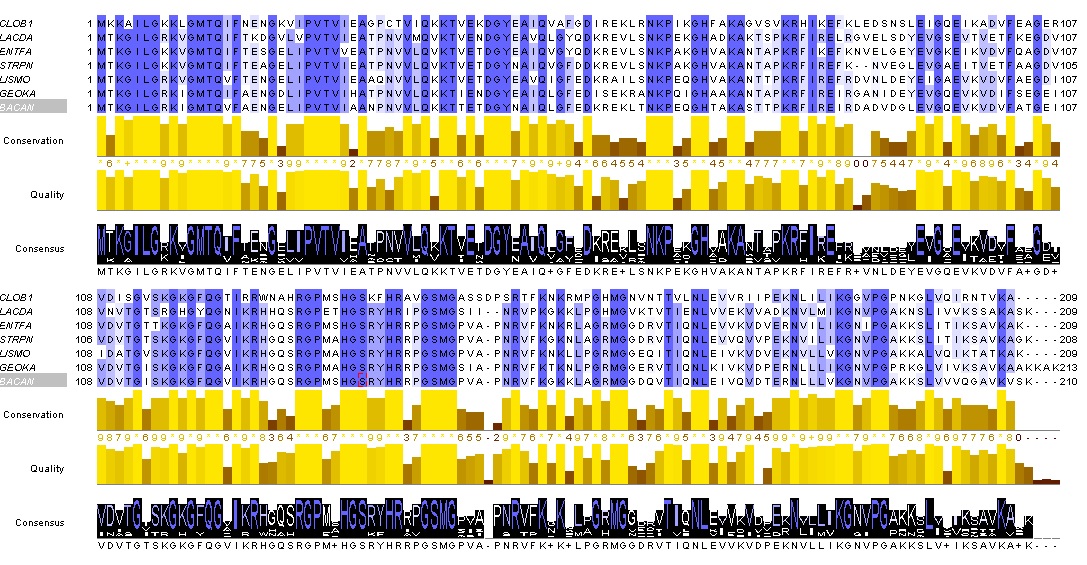 |
Рис. 1 Выравнивание рибосомальных белков RL3 из семи видов бактерий. |
Поиск диагностических позиций в выравнивании
Диагностическая позиция - это такой номер а/к в последовательности белка, что то, какая а/к стоит в данном месте, коррелирует с систематическим положением организма (на определённом уровне таксонов).- Clostridia/Bacilli - позиции 4 и 138
- Bacillales/Lactobacillales - позиция 89
Сравнение топологии полученных деревьев с топологией правильного дерева
C помощью четырёх различных методов, доступных в программе Jalview, были построены филогенетические деревья. Они приведены в таблице ниже. Далее дано сравнение топологий (наличия/отсутствия тех или иных ветвей в дереве).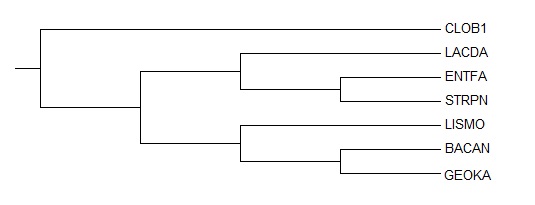 |
Правильное филогенетическое дерево для семи видов бактерий, полученное при выполнении первого практикума |
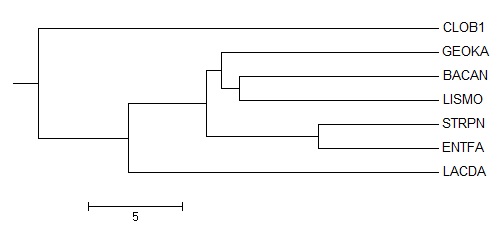 |
Дерево Average distance using % identity |
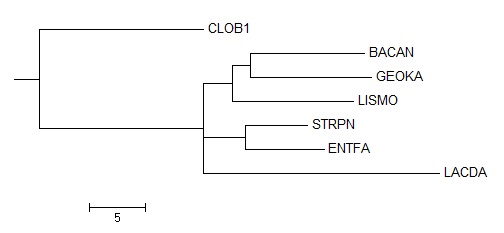 |
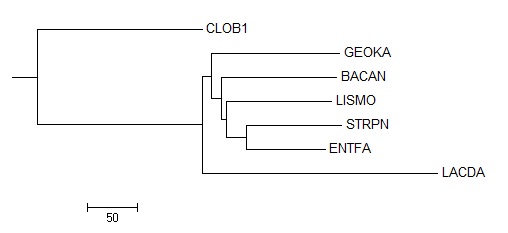
Дерево Neighbour joining using % identity |
 |
Дерево Average distance using Blosum62 |
 |
Дерево Neighbour joining using BLOSUM62 |
В таблице ниже приведены результаты по наличию ветвей во всех рассматриваемых в данном практикуме деревьях. В каждом дереве присутствует по 4 ветви, кроме дерева, построенного методом Neighbour joining using % identity, так как оно получилось не бинарным. Стоит отметить, что ни один из использованных методов не смог дать правильной исходной конфигурации дерева, что в целом объясняет такое разнообразие методов. Большинство методов верно воспроизвело две из четырёх ветвей исходного дерева.
В качестве отдельных наблюдений отмечу: (i) ветвь {STRPN+ENTFA}&{others} встречается во всех построенных деревьях, что говорит о том, что последовательности их белков настолько сходны, что вне зависимости от метода попадают в одну кладу,(ii) вероятно такой разброс в полученных результатах связан с тем, что выравнивание строилось на основе одного белка, в то время как в настоящих филогенетических исследованиях берутся целые семейства белков и РНК.
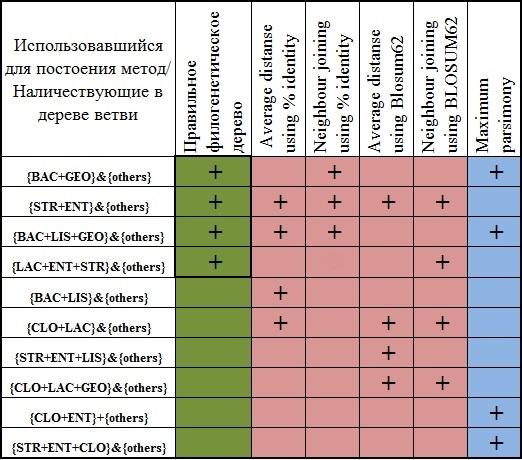 |
Рис.3 Сравнительная таблица топологии рассмотренных деревьев. По столбцам расположены пять методов, которые использовались для построения деревьев, а также добавлен столбец, соответствующий исходному правильному дереву. По строкам расположены различные разбиения множества листьев (ветви), встреченные на различных деревьях. |
Реконструкция дерева методом Maximum Parsimony с помощью программы Mega
С помощью программы MEGA по данным исходного выравнивания было построено филогенетическое дерево методом максимальной экономии и укоренено по вручную. сравнение топологии для этого и правильного деревьев приведено в таблице выше.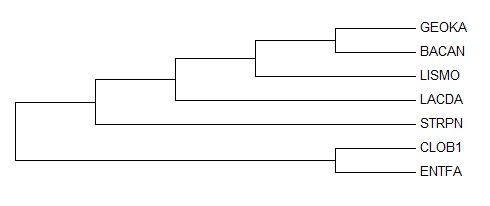 |
Рис.4 Филогенетическое дерево, полученное методом максимальной экономии (maximum parsimony) |
Дата последнего обновления: 23.02.2013
© Dmitry Travin, 2014