Совмещение структуры белка и его гомологов
При помощи сервиса PDBeFold были найдены четыре гомолога белка 3QTP. Эти структуры также принадлежат енолазам, но из других организмов: 5bof
Staphilococcus aureus, 1pdy
Homarus gammarus, 3b97
Homo sapiens и 4a3r
Bacillus subtilis. Для пяти структур было построено пространственное выравнивание (см. Рис. 1 и Рис. 2).
 |
 |
 |
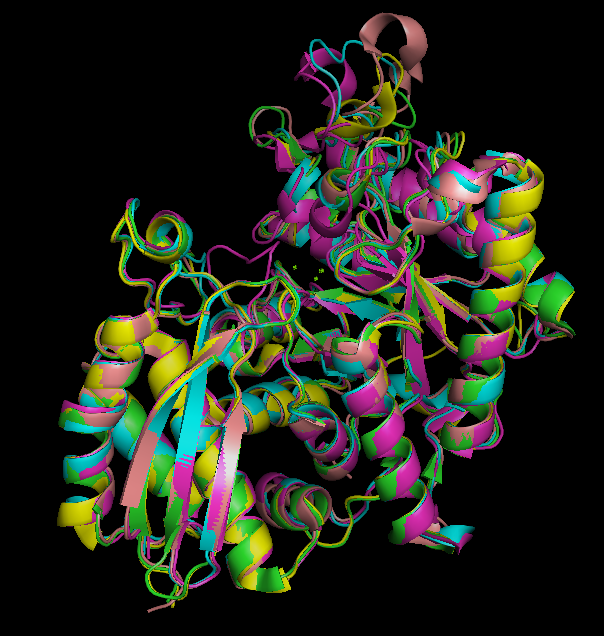
Рис. 1 Структурное выравнивание 3QTP (розовый) с четырьми его структурными гомологами: 1pdy (голубой), 4a3r (зелёный), 3b97 (магента), 5bof (жёлтый). Визуализация Pymol. |
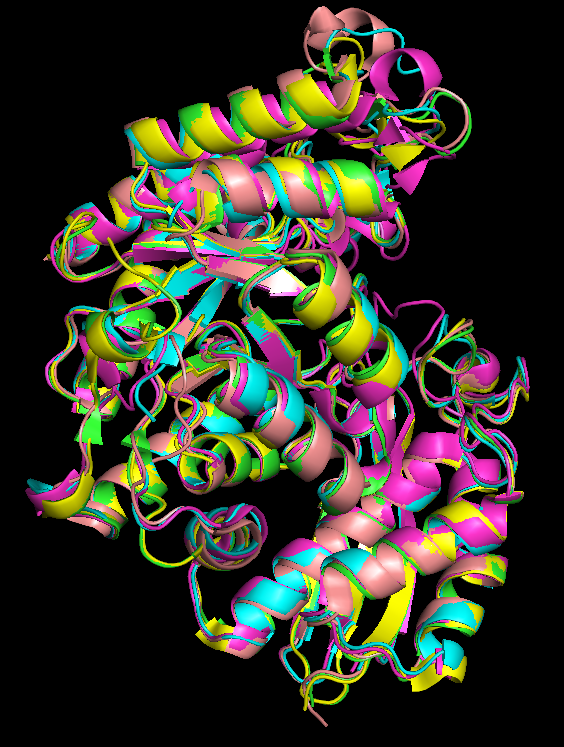
Рис. 2 Структурное выравнивание 3QTP и его гомологов, выполненное при помощи PDBeFold и визуализированное при помощи Jmol. |
По
ССЫЛКЕ доступно выравнивание последовательностей, полученное из совмещения вторичных структур белка и его гомологов. Кроме того, было получено выравнивание последовательностей белков при помощи алгоритма MUSCLE.
 |
Рис. 3 Выравнивание последовательностей белка и его гомологов, полученное применением алгоритма MUSCLE. Раскраска по консервативности позиций. |
Ситуаций, когда остатки выровнены структурно, но не выровнены по последовательности, найдено не было. Зато были найдены контрпримеры, когда структурно не выровненные участки выровнены, исходя из последовательности (см. Рис. 4).
 |
 |
Рис. 4 Ситуации несовпадения структурного выравнивания и выравнивания, основанного на последовательностях. |
Рассмотрим для первого случая то, как же это выглядит, если выделить выровненные по последовательности глицины на структуре (см. Рис. 5). Как видно из рисунка, все они находятся сравнительно рядом с друг другом на конце альфа-спирали. Мы знаем, что глицин, это аминокислота, которая "любит" быть на конце спирали в связи с облегчённым вращением по углам, которое благоприятно при переходе от регулярного участка к нерегулярному. Мне в данном случае сложно сказать, являются ли эти остатки гомологичными и стоит ли их выравнивать.
 |
Рис. 5 Ситуация несовпадения структурного выравнивания и выравнивания, основанного на последовательностях. Показаны (красным) пять глицинов на конце альфа-спирали, не выровненных по структуре, но выровненных по последовательности. |
Поиск по сходству структур в PDBeFold
Был осуществлён поиск по сходству с помощью сервиса PDBeFold для домена 1b0m A:203-315. Выдача была отсортирована в соответствии с RMSD. Всего нашлось 43 сходных структуры, но исходной (по которой осуществлялся поиск) среди них обнаружено не было. Выдача доступна по
ССЫЛКЕ. Далее, чтобы разобраться в происходящем, я отправил запрос поиска этого домена в его же полной структуре (1b0m A:203-315 против 1b0m) и получил процент sse (общих вторичных структур) равным 16%. Таким образом, я уверен, если бы я сделал поиск не с 70% минимального сходства вторичной структуры, а с 15%, а потом отсортировал по RMSD, то вся структура 1b0m была бы первой в списке. Делать я этого не стал в целях сбережения времени и интернет трафика.
Совмещение по заданному выравниванию
Из PDB структуры 1OGA были получены отдельные PDB-файлы, соответствующие доменам альфа-цепи (цепь D:118-202) и бета-цепи (цепь E:119-245). Файлы доступны по ссылкам:
D:118-202 и
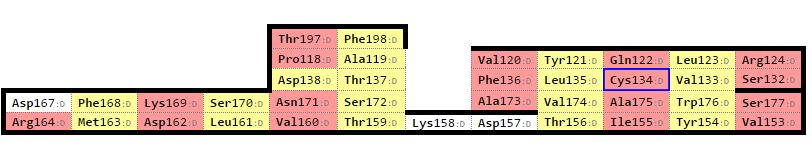
E:119-245. Выбранные участки на общей структуре белка показаны на рисунке 6. Затем для этих двух файлов были получены карты бета листов при помощи программы SheeP. Они приведены на рисунке 7. Верхнему листу в цепи бета соответствует лист из альфа.
 |
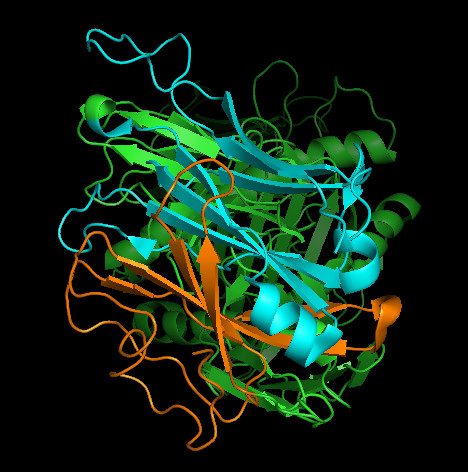 |
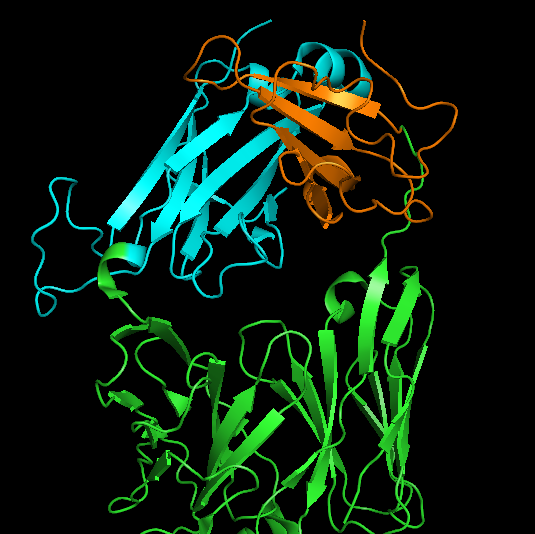
Рис. 6 Структура выбранных участков (вид сбоку и вид сверху), визуально различимы три бета-листа. |
 |
 |
Рис. 7 Карты бета листов полученные программой Sheep для двух участков из алфа- (слева) и бета-цепи константного домена T-клеточного рецептора. Синими рамочками отмечены консервативные остатки цистеинов. |
Далее было проведено выравнивание двух выделенных доменов с помощью следующего скрипта для Pymol:
cd D:\Bioinfa\pr_8
load ALFA.pdb
load BETA.pdb
select 31, ALFA and (resi 133-135 or resi 122 or resi 175) and name ca
select 32, BETA and (resi 144-146 or resi 127 or resi 192) and name ca
pair_fit 31, 32
show cartoon, all
hide sticks, all
hide lines, all
Как видно из кода, в качестве основы для выравнивания берутся
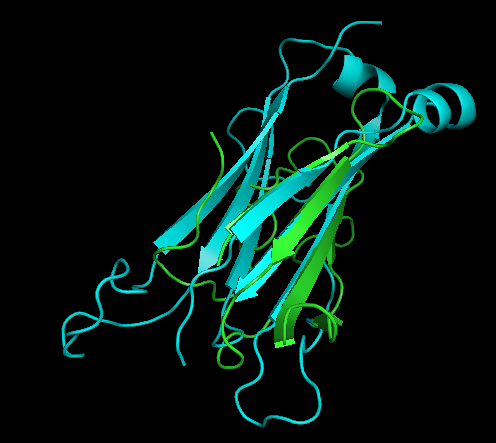
5 аминокислотных остатков: консервативные цистеины двух соответствующих бета-листов, две соседних аминокислоты того же тяжа для каждого из цистеинов и 2 аминокислотных остатка того же гребня, что и цистеин, но в других тяжах. Применение именно такого набора из пяти точек пространства позволяет нам задать топологию тяжа в двух направлениях, поскольку один тяж задаёт её не совсем точно и не отражает "чипсовидной" формы листа. Полученное пространственное наложение структур показано на рисунке 8. Видно, что бета-листы хорошо соответствуют друг другу, в то время как общая топология участков не очень совпадает, хотя можно подметить, что есть нечто общее в ходе полипептидной цепи. Файл с полученным совмещением доступен по
ССЫЛКЕ.
 |
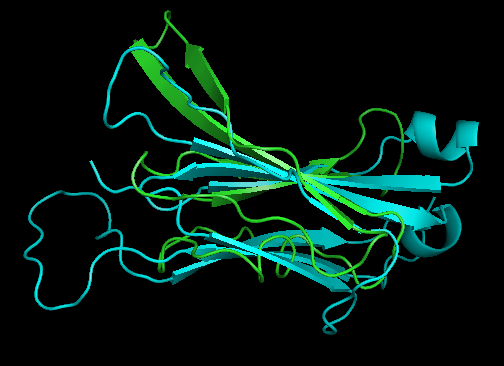 |
Рис. 8 Совмещение структур двух участков цепей Т-клеточного рецептора, построенное на основе совмещения консервативного цистеина. Показано два вида. Цепи отмечены разными цветами (голубым - beta, зелёным - alpha). |