Часть I: подготовка чтений.
fastqc chr21.fastqВывод программы:
Started analysis of chr21.fastq Approx 10% complete for chr21.fastq Approx 20% complete for chr21.fastq Approx 35% complete for chr21.fastq Approx 45% complete for chr21.fastq Approx 60% complete for chr21.fastq Approx 70% complete for chr21.fastq Approx 85% complete for chr21.fastq Approx 95% complete for chr21.fastq Analysis complete for chr21.fastqВ результате были получены 2 файла:chr21_fastqc.zip и chr21_fastqc.html.
Результаты в формате .html: chr21_fastqc.html.
Далее была произведена очистка чтений с помощью программы Trimmomatic. При этом программа Trimmomatic была запущена с помощью команды:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr21.fastq c.fastq TRAILING:20 MINLEN:50Вывод программы:
TrimmomaticSE: Started with arguments: -phred33 chr21.fastq c.fastq TRAILING:20 MINLEN:50 Automatically using 8 threads Input Reads: 8158 Surviving: 7858 (96,32%) Dropped: 300 (3,68%) TrimmomaticSE: Completed successfullyВ результате был получен файл c.fastq. Далее был произведен анализ качества чтений файла c.fastq. Программа FastQC была запущена через командную строку на kodomo:
fastqc c.fastqВывод программы:
Started analysis of c.fastq Approx 10% complete for c.fastq Approx 25% complete for c.fastq Approx 35% complete for c.fastq Approx 50% complete for c.fastq Approx 60% complete for c.fastq Approx 75% complete for c.fastq Approx 85% complete for c.fastq Analysis complete for c.fastqВ результате были получены 2 файла:c_fastqc.zip и c_fastqc.html.
Результаты в формате .html: c_fastqc.html.
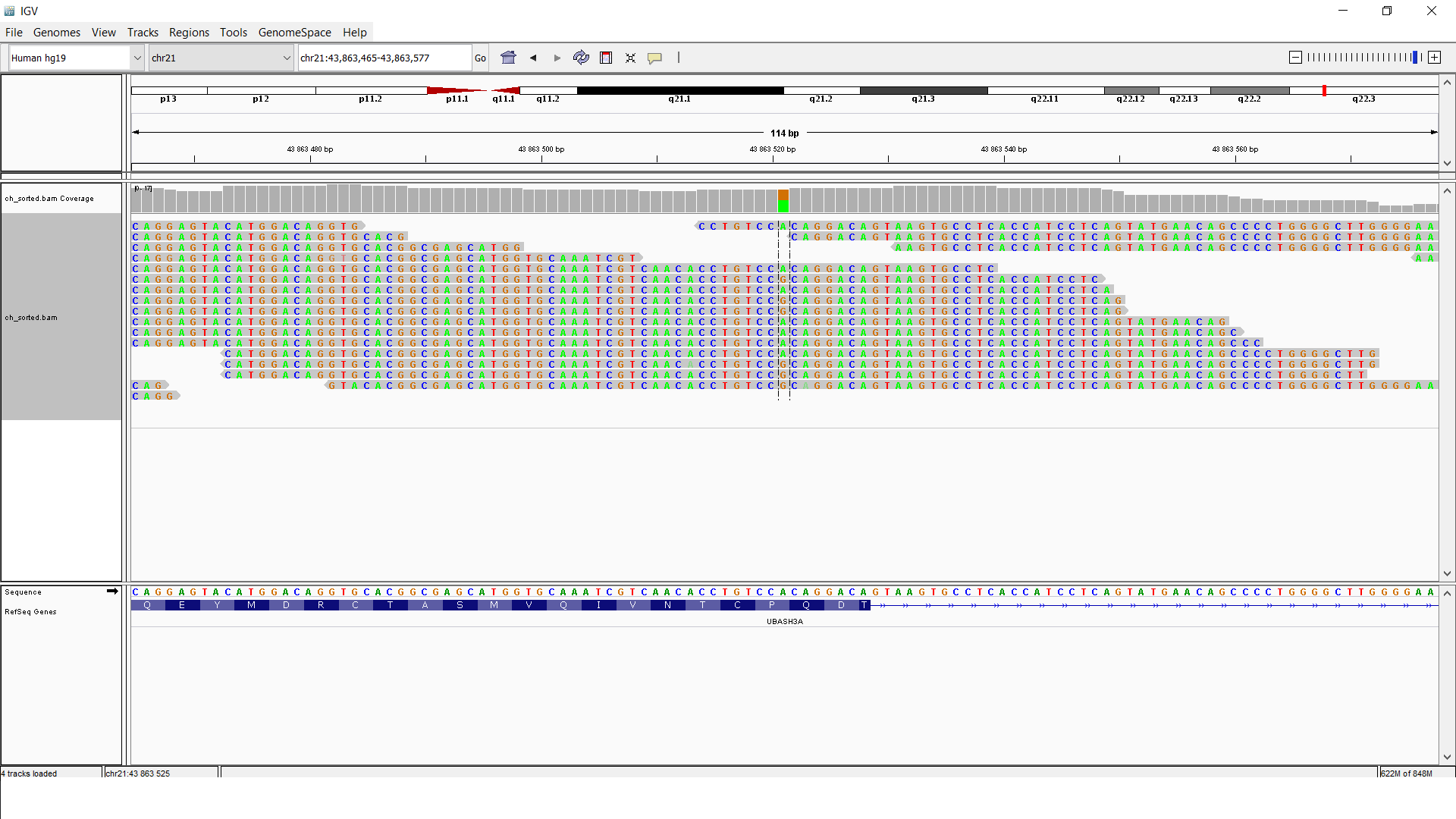
Качество прочтения нуклеотидов
| До чистки Trimmomatic |
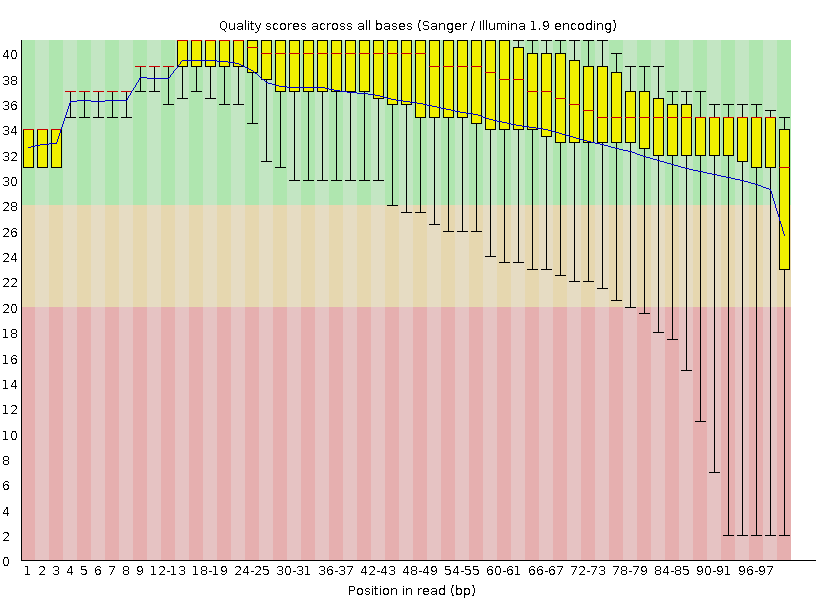 |
| После чистки Trimmomatic |
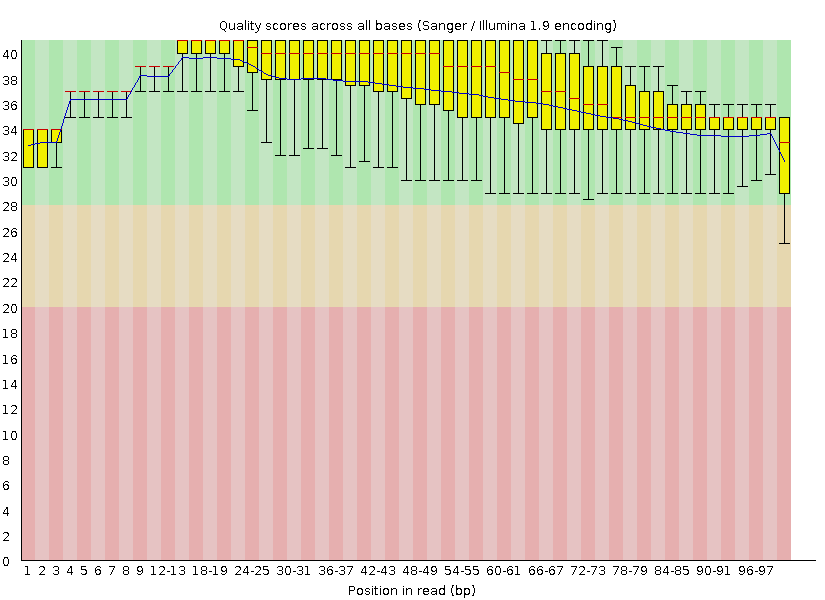 |
Как видно из приведенных рисунков, качество прочтений после работы программы Trimmomatic повысилось, и теперь почти все позиции нуклеотидов прочтены хорошо , так как почти попадают в зелёную область гистограммы, что означает quality от 28 и выше. До работы программы Trimmomatic качество некоторых прочтений было неудовлетворительным, так как онипопадали в красную область. Также можно заметить, что наибольшее повышение качества произошло на конце ридов, поскольку программа Trimmomatic обрезала концевые нуклеотиды, имевшие качество ниже 20.
Распределение качества прочтений
| До чистки Trimmomatic |
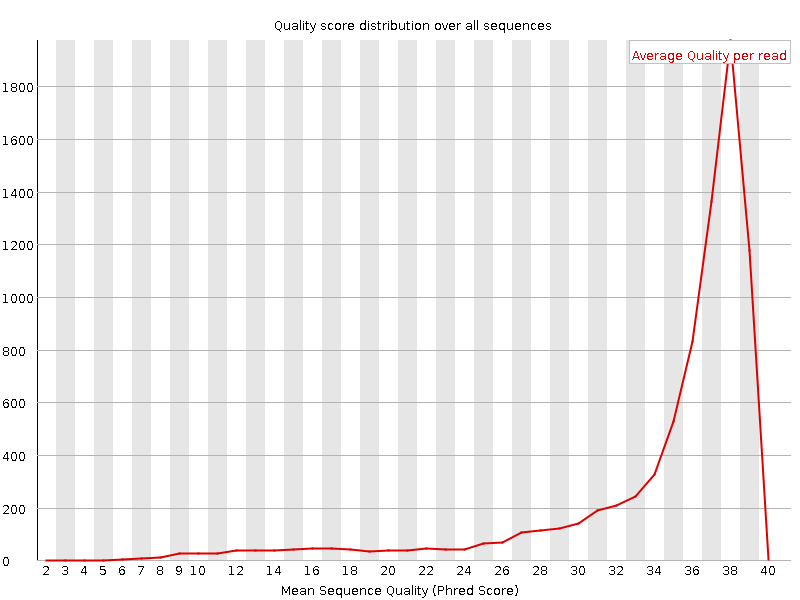 |
| После чистки Trimmomatic |
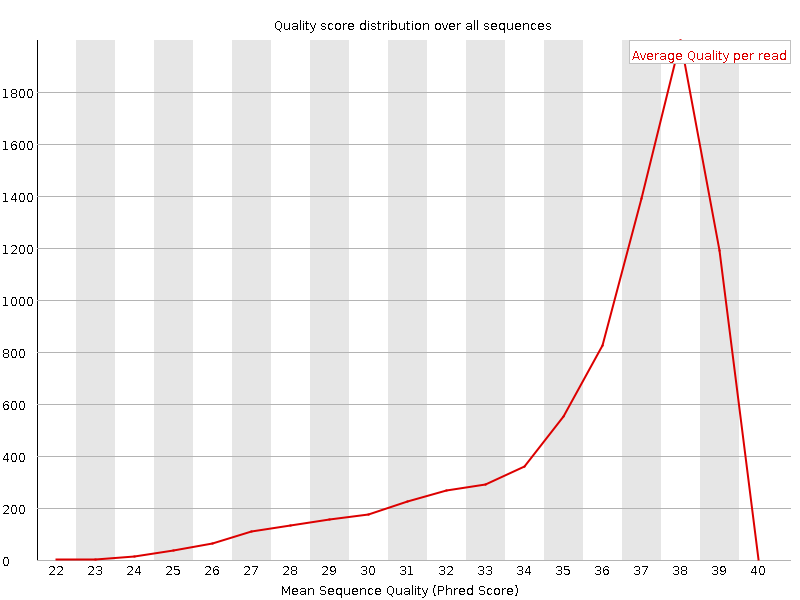 |
На этих рисунках представлены графики из файлов chr21_fastqc.html и c_fastqc.html выданных FastQC, на которых представлено распределение качества по последовательностям в целом. Нужно заметить, у 2 рисунков отличается шкала по горизонтальной оси, на которой отражено среднее качество последовательности. На рисунке 1, отображающем результаты анализа качества чтений до чистки программой Trimmomatic шкала начинается с 2, а рисунке 2, отображающем результаты анализа качества чтений после чистки программой Trimmomaticшкала начинается с 22. Это демонтрирует работу программы Trimmomatic, поскольку как можно заметить из приведенных графиков, программа Trimmomatic действительно удаляет последовательности с низким (меньше 20) качеством (phred-score).
Команды.
| Номер | Команда | Действие |
| 1 | fastqc chr21.fastq | Запуск FastQC для анализа качества чтений файла cрк21.fastq. На выходе — html-страница с результатами(файл chr21_fastqc.html) и архив в формате .zip. |
| 2 | java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr21.fastq c.fastq TRAILING:20 MINLEN:50 | Запуск Trimmomatic в режиме SE (single-ends) для того, чтобы улучшить качества одноконцевых прочтений. Производится очистка чтений. В процессе очистки чтений отрезаются с конца каждого чтения нуклеотиды с качеством ниже 20, оставляются только чтения длиной не меньше 50 нуклеотидов.
-phred33 задает формат, в котором задаётся качество в fastq-файле TRAILING:20 удаляет с конца каждого чтения нуклеотиды с качеством ниже 20 MINLEN:50 убирает чтения с длиной менее, чем 50 нуклеотидов. |
| 3 | fastqc c.fastq | Запуск программы FastQC для анализа качества чтений файла c.fastq. На выходе — html-страница с результатами(файл c_fastqc.html) и архив в формате .zip. |
Количество и длина чтений
 |
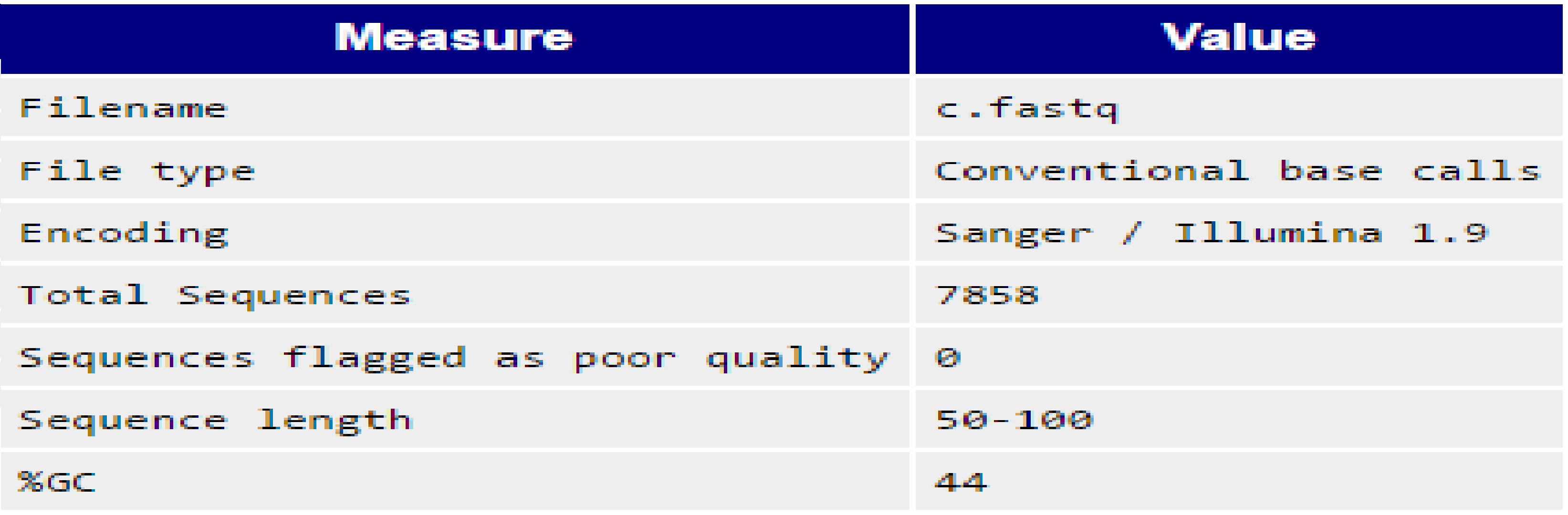 |
Из 2 рисунков с общей статистикой можно сделать выводы о числе и длине чтений до и после чистки:
| Длина чтений | Количество чтений | |
| До очистки | 8158 | 30-100 |
| После очистки | 7858 | 50-100 |
Из приведенной таблицы видно, что в начале было 8158 чтений с длиной от 30 до 100, после обработки их число уменьшилось до 7858, причём остались только риды с длиной от 50 до 100.
Часть II: картирование чтений.
bwa index chr21.fastaВывод программы:
[bwa_index] Pack FASTA... 0.88 sec [bwa_index] Construct BWT for the packed sequence... [BWTIncCreate] textLength=96259790, availableWord=18772880 [BWTIncConstructFromPacked] 10 iterations done. 30966126 characters processed. [BWTIncConstructFromPacked] 20 iterations done. 57205886 characters processed. [BWTIncConstructFromPacked] 30 iterations done. 80523822 characters processed. [bwt_gen] Finished constructing BWT in 38 iterations. [bwa_index] 121.17 seconds elapse. [bwa_index] Update BWT... 0.72 sec [bwa_index] Pack forward-only FASTA... 0.67 sec [bwa_index] Construct SA from BWT and Occ... 25.43 sec [main] Version: 0.7.10-r789 [main] CMD: bwa index chr21.fasta [main] Real time: 314.423 sec; CPU: 148.880 secДалее было построено выравнивание прочтений и референса в формате .sam, при этом был использован алгоритм mem и команда bwa mem. Для построения выравнивания прочтения и референса была использована команда:
bwa mem chr21.fasta c.fastq > ch.samВывод программы:
[M::main_mem] read 7858 sequences (769104 bp)... [M::mem_process_seqs] Processed 7858 reads in 0.436 CPU sec, 1.035 real sec [main] Version: 0.7.10-r789 [main] CMD: bwa mem chr21.fasta c.fastq [main] Real time: 3.481 sec; CPU: 0.708 secВ результате был получен файл ch.sam с выравниванием прочтений и референса. Далее выравнивание чтений с референсом было переведено в бинарный формат .bam. При этом был использован пакет samtools, команда view: samtools view. Чтобы узнать необходимую опцию команды samtools view был вызван help этой команды. Дляэтого использована команда:
Usage: samtools view [options]Для перевода выравнивания чтений с референсом было в бинарный формат .bam нужно использовать опции -b и -o.| | [region ...] Options: -b output BAM -C output CRAM (requires -T) -1 use fast BAM compression (implies -b) -u uncompressed BAM output (implies -b) -h include header in SAM output -H print SAM header only (no alignments) -c print only the count of matching records -o FILE output file name [stdout] -U FILE output reads not selected by filters to FILE [null] -t FILE FILE listing reference names and lengths (see long help) [null] -T FILE reference sequence FASTA FILE [null] -L FILE only include reads overlapping this BED FILE [null] -r STR only include reads in read group STR [null] -R FILE only include reads with read group listed in FILE [null] -q INT only include reads with mapping quality >= INT [0] -l STR only include reads in library STR [null] -m INT only include reads with number of CIGAR operations consuming query sequence >= INT [0] -f INT only include reads with all bits set in INT set in FLAG [0] -F INT only include reads with none of the bits set in INT set in FLAG [0] -x STR read tag to strip (repeatable) [null] -B collapse the backward CIGAR operation -s FLOAT integer part sets seed of random number generator [0]; rest sets fraction of templates to subsample [no subsampling] -@ INT number of BAM compression threads [0] -? print long help, including note about region specification -S ignored (input format is auto-detected)
Для Для перевода выравнивания чтений с референсом было в бинарный формат .bam была использована команда:
samtools view ch.sam -b -o ch.bamВ результате был получен файл ch.bam. Затем выравнивание чтений с референсом (получившийся после картирования .bam файл) был отсортирован по координате начала чтения в референсе. Для того, чтобы выяснить необходимые опции команды был вызван help. Выдача help:
Usage: samtools sort [options...] [in.bam]
Options:
-l INT Set compression level, from 0 (uncompressed) to 9 (best)
-m INT Set maximum memory per thread; suffix K/M/G recognized [768M]
-n Sort by read name
-o FILE Write final output to FILE rather than standard output
-O FORMAT Write output as FORMAT ('sam'/'bam'/'cram') (either -O or
-T PREFIX Write temporary files to PREFIX.nnnn.bam -T is required)
-@ INT Set number of sorting and compression threads [1]
Legacy usage: samtools sort [options...]
Options:
-f Use as full final filename rather than prefix
-o Write final output to stdout rather than .bam
-l,m,n,@ Similar to corresponding options above
Для сортировки выравнивания была использована команда:
samtools sort -T /tmp/ -o ch_sorted.bam ch.bamВ результате был получен файл:ch_sorted.bam . Затем полученный файл был проиндексирован командой:
samtools index ch_sorted.bamВ результате был получен файл ch_sorted.bam.bai. Далее для получения информации о количестве картированных прочтений была использована команда:
samtools idxstats ch_sorted.bamВыдача программы:
chr21 48129895 7858 0 * 0 0 1
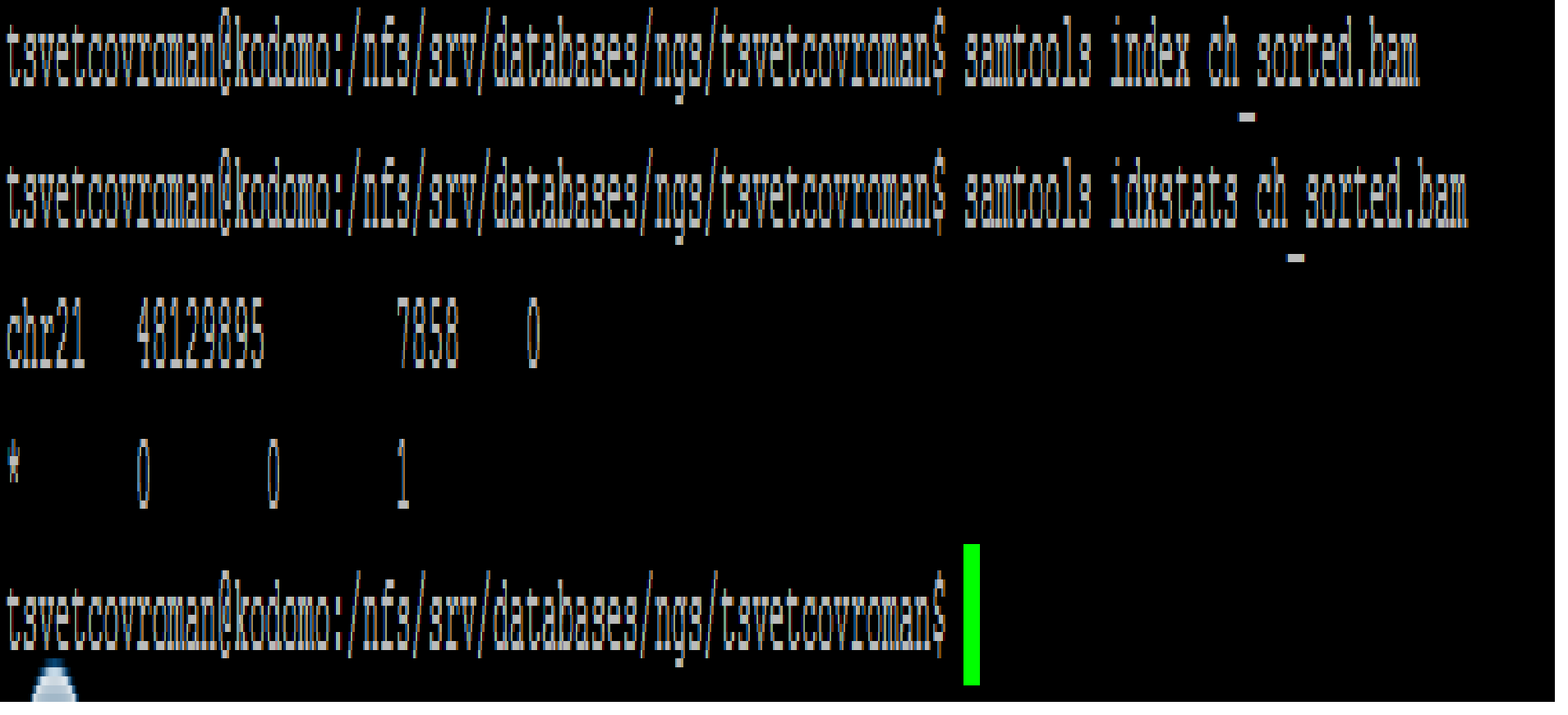
Рисунок 1.Выдача программы samtools idxstats ch_sorted.bam.
Из выдачи программы можно узнать, что на референсную последовательность картировались все 7858 прочтений, оставшиеся после улучшения качества с помощью программы Trimmomatic.
Часть III: Анализ SNP.
samtools mpileup -uf chr21.fasta ch_sorted.bam > snp.bcfВыдача команды:
[fai_load] build FASTA index. [mpileup] 1 samples in 1 input filesВ результате был получен файл snp.bcf. Далее был создан файл со списком отличий между референсом и чтениями в формате .vcf . Для этого была использована команда:Set max per-file depth to 8000
bcftools call -cv snp.bcf > snp.vcfВ результате был получен файл snp.vcf.
В файле записано 6 инделей и 82 замен. Данные полиморфизмы довольно низкого качества: медианное качество 11,34 и среднее 57,81.
Ссылка на файл с расчетами:s.xlsx. Примеры полиморфизмов: