1. Анализ качества чтений
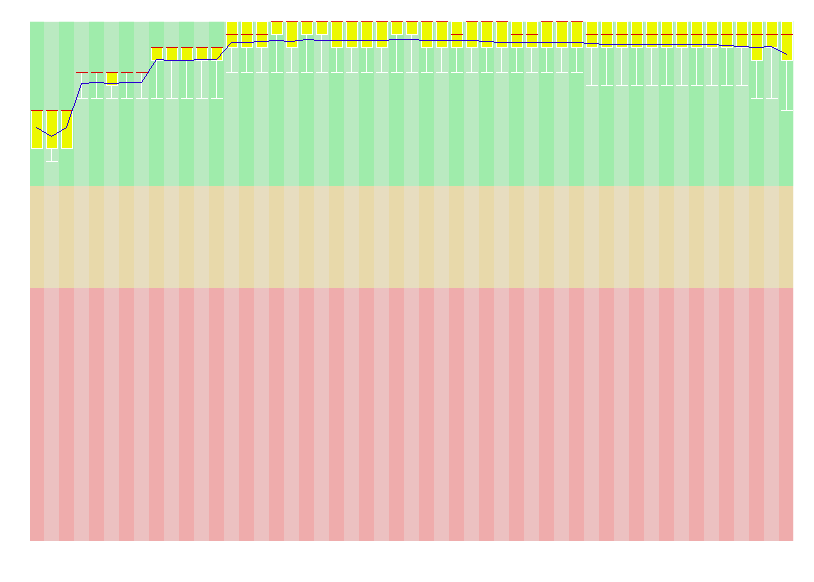
Рисунок 1. Изменение качества чтений до чистки
Далее было необходимо очистить чтения от участков плохого качества. Это можно сделать с помощью программы Trimmomatic:
$ java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr17.1.fastq rna_trim.fastq TRAILING:20 MINLEN:50
Trimmomatic оставила чтения длиной не меньше 50 (MINLEN:50) и с конца каждого такого чтения удалила нуклеотиды с качеством ниже 20 (TRAILING:20). После чистки число чтений сократилось с 10407 до 10355 (99,50% от общего числа). Очищенные чтения подали на вход программе FastQC:
$ fastqc rna_trim.fastq
На рисунке 2 представлена диаграмма из отчета, аналогичная той, что была получена для неочищенных чтений:
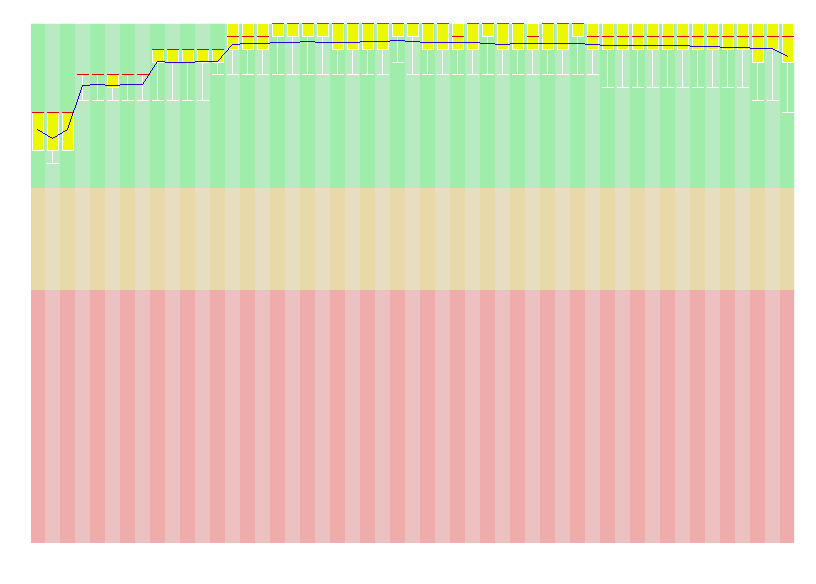
Рисунок 2. Изменение качества чтений после чистки
2. Картирование чтений
Для картирования вновь воспользуемся программой Hisat2 и уже проиндексированой в прошлом практикуме референсной последовательностью семнадцатой хромосомы. Однако в этот раз мы не будем использовать параметр --no-spliced-alignment, так как при работе с транскриптомом отсеквениованный нами участок в геноме может соответствовать двум экзонам, разделенных интроном.
$ hisat2 -x ../indexed_chr17/chr17 -U rna_trim.fastq -S rna_align.sam --no-softclip 2> rna.log
3. Анализ выравнивания
| $ samtools view rna_align.sam -bo rna_align.bam | Переводит выравнивание чтений с референсом в бинарный формат .bam |
| $ samtools sort rna_align.bam sorted | Сортирует выравнивание чтений с референсом по координате в референсе начала чтения |
| $ samtools index sorted.bam | Индексирует отсортированный .bam файл |
Вывод программы Hisat2 выглядит следующим образом:
10355 reads; of these:
10355 (100.00%) were unpaired; of these:
326 (3.15%) aligned 0 times
10029 (96.85%) aligned exactly 1 time
0 (0.00%) aligned >1 times
96.85% overall alignment rate
Итого, число чтений, картированных на референсную хромосому: 10355
Число не картированных чтений: 326
4. Подсчет чтений
Программа htseq-count позволяет узнать число ридов, наложившихся на референсную последовательность. Ниже представлены некоторые ее опции и команда вызова:
- -f <format>: определяет формат входного файла: sam или bam.
- -s <yes/no/reverse>: yes указывает на то, что рид соответствует той же цепи, что и референс; no отменяет это условие (default: yes).
- -i <id attribute>: GFF атрибут, используемый в качестве feature-id. Для RNA-Seq это gene_id.
- -m <mode>: В зависимости от параметра (union, intersection-strict, intersection-nonempty), определяет, что делать с перекрывшими несколько features ридами. Принцип работы каждого из параметров хорошо описан в документации HTSeq 0.8.0.
$ htseq-count -f bam sorted.bam -m union -i gene_id -s no annotation.gtf > htseq_count
Для поиска не содержащих "0" строк используем grep:
$ grep -wv 0 htseq_count
Из вывода grep'a (представлен справа) можно заключить, что 414 ридов не легли в границы генов, 906 попали в границы сразу нескольких генов, но не были присвоены ни одному из них, и 326 не соответсвтуют ни одному выравниванию.
ENSG00000108424.5 8708 ENSG00000263766.1 1 __no_feature 414 __ambiguous 906 __not_aligned 326
ENSG00000108424.5 (KPNB1) - ген белка импортин-бета-1, автономно, в качестве рецептора импорта ядерных белков, или в комплексе с другими белками-транспортинами принимающего участие в транспорте белков через ядерные поры.
ENSG00000264558.1 кодирует antisense транскрипт для KPNB1 - олигонуклеотид, который, гибридизуясь с геном KPNB1, препятсвует его трансляции в белок.
При запуске htseq-count с параметром -m intersection-strict количество чтений, относящихся к ENSG00000264558.1, сократилось до 601. Ссылаясь на документацию, это можно объяснить тем, что исключенные риды могли находиться либо на краях гена, либо между двумя экзонами.
Аналогично прошлому практикуму, с помощью программы FastQC проанализируем качество чтений, полученных в результате секвенирования транскриптома человека.
$ fastqc chr17.1.fastq
В результате ее работы был получен .html файл с отчетом. На рисунке 1 в виде боксплота представлено изменение качества чтений на каждый нуклеотид.