Сборка генома de novo
В данном практикуме предлагалось провести сборку генома бактерии Buchnera aphidicola str. Tuc7 de novo на основе коротких (39 нуклеотидов) одноконцевых чтений, полученных по технологии Illumina. AC ДНК-чтений в моём случае — SRR4240356. Поиск архива с чтениями осуществлялся на сайте ENA. Затем с помомью команды wget этот архив скачивался в рабочую директорию. Команда для скачивания:
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/006/SRR4240356/SRR4240356.fastq.gz
Подготовка чтений программой Trimmomatic
Перед непосредственной сборкой на основе чтений требовалось удалить с их концов возможные остатки адаптеров с помощью программы TrimmomaticSE (т.к. работаем с одноконцевыми прочтениями). Для этого был создан файл в формате FASTA adapters.fa, содержащий все последовательности (и названия) адаптеров для Illumina, находящиеся в директории /mnt/scratch/NGS/adapters. По результатам работы программы выяснилось, что 2.18% последовательностей чтений оказались остатками адаптеров (на вход было подано 7511529 чтений, из них программа TrimmomaticSE удалила 163510). Команда, с помощью которой была запущена программа:
TrimmomaticSE SRR4240356.fastq.gz lane_forward.fq.gz ILLUMINACLIP:adapters.fa:2:7:7Далее также с помощью TrimmomaticSE с правого конца чтений были удалены нуклеотиды с качеством ниже 20 и оставлены только те чтения, длина которых не меньше 32 нуклеотидов. В результате было удалено ещё 294834 чтения (4.01% от оставшихся после удаления адаптеров). Команда, с помощью которой была запущена программа:
TrimmomaticSE lane_forward.fq.gz lane_forward_itog.fq.gz TRAILING:20 MINLEN:32Размеры файлов до и после очистки можно узнать с помощью команды du -h:
du -h SRR4240356.fastq.gz
du -h lane_forward.fq.gz
du -h lane_forward_itog.fq.gzИсходный файл с неочищенными чтениями SRR4240356.fastq.gz весил 167 Мб, файл с удалёнными адаптерами lane_forward.fq.qz — 164 Мб и файл с отфильтрованными по длине и качеству концевых нуклеотидов чтений lane_forward_itog.fq.gz — 155 Мб.
Создание k-меров с помощью программы velveth
Далее была использована программа velveth, которая создаёт k-меры (слова из нуклеотидов длины k) для их последующего объединения в контиги. В данном случае длина k-меров была максимально возможной для наших ДНК-чтений и составила 31 нуклеотид. Чтения в нашем случае были короткие (опция -short) и не парные, формат входного файла — fastq.gz (опция -fastq.gz), Pr_14_assembly_velveth — имя поддиректории, куда сохраняются выходные файлы программы. Команда, с помощью которой запускалась программа:
velveth Pr14_assembly_velveth 31 -short -fastq.gz lane_forward_itog.fq.gz
Сборка на основе k-меров с помощью программы velvetg
Затем использовалась программа velvetg, предназначенная для сборки контигов на основе k-меров (в нашем случае k = 31). Для её запуска требовалось указать поддиректорию, куда были сохранены выходные файлы программы velveth. Реализация осуществлялась с помощью следующих команд:
cd Pr14_assembly_velveth/
velvetg .На выходе получился граф де Брёйна, состоящий из 286 вершин. Параметр N50 собранных контигов составил 65554. Далее была написана команда, выводящая названия (номера вершин), длины и покрытия трёх самых длинных контигов:
grep '^>' contigs.fa | tr '_' '\t' | cut -f2,4,6 | sort -k2,2 -n -r | head -3Результаты выдачи представлены в Таблице 1.
| Название (номер вершины) контига |
Длина в нуклеотидах | Покрытие |
|---|---|---|
| 8 | 111962 | 38.660198 |
| 6 | 107488 | 34.174030 |
| 10 | 80939 | 37.524174 |
Затем необходимо было определить, присутствуют ли в данной сборке контиги с аномально большим или аномально малым покрытием, т.е. таким, которое отличается от медианного значения более чем в 5 раз. Для этого все контиги были отсортированы по длине (чтобы более наглядно показать аномальные значения, хотя это необязательно) и записаны в соответствующий tsv-файл, который затем был преобразован в формат csv и импортирован в Excel, в котором и проводился расчёт медианного значения покрытия.
grep '^>' contigs.fa | tr '_' '\t' | cut -f2,4,6 | sort -k3,3 -n -r > contigs_sorted_cov.tsv
tr '\t' ',' < contigs_sorted_cov.tsv | cat > contigs_sorted_cov.csvПосле проведения анализа в Excel, нетрудно заметить, что, например, аномально большим покрытием обладают контиги 27 (длина 282), 17 (длина 950), 14 (длина 934), а аномально малым — 74 (длина 31) и 123 (длина 91). Ссылка на Excel-файл.
Анализ полученных результатов сборки генома
В конце проводилось сравнение каждого из трёх наиболее длинных контигов с хромосомой другого штамма бактерии Buchnera aphidicola — strain F009
(см. Рис.7., Рис.8., Рис.9.).
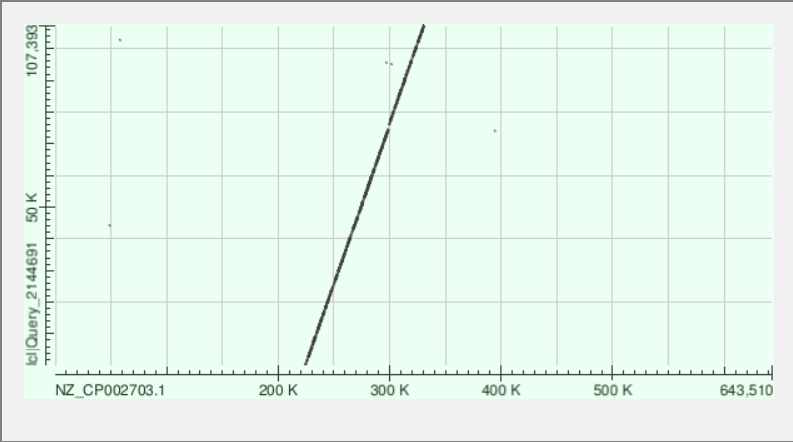
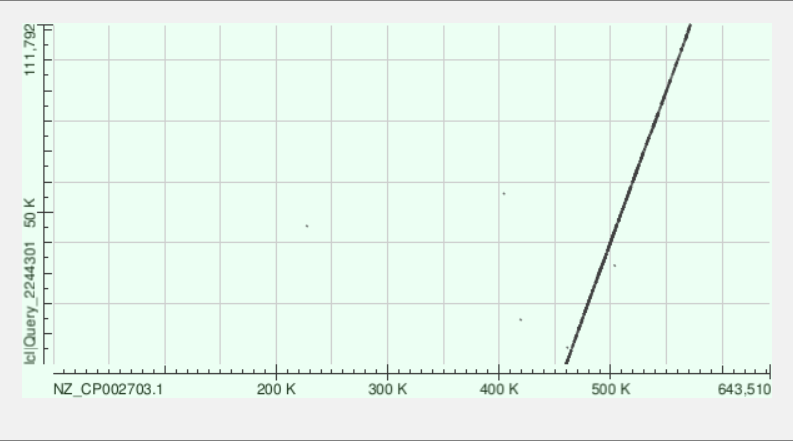
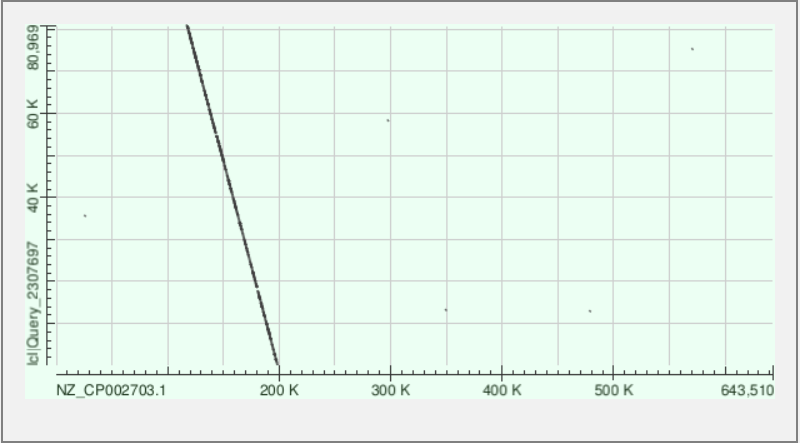
Для 6-го контига получилось 23 выравнивания, в основном длинной от 1 до 19 Kb. Суммарная длина выравнивания составила 105302 нуклеотида. Доля контига, выравненная на геном, составила 105302/107518 * 100 = 97.94%. Участок хромосомы, куда произошло выравнивание — с 224334 по 330746 нуклеотиды (здесь и далее учитывались только выравнивания с длинной больше 200). Число однонуклеотидных несоответствий (мисматчей) составило 17193, число гэпов — 1597 (см. Excel-файл и текстовый файл выдачи BLAST).
Для 8-го контига получилось 24 выравнивания, в основном длинной от 1 до 32 Kb. Суммарная длина выравнивания составила 110199 нуклеотидов. Доля контига, выравненная на геном, составила 110199/111992 * 100 = 98.40%. Участок хромосомы, куда произошло выравнивание контига — с 460068 по 571665 нуклеотиды. Число мисматчей — 17083, число гэпов — 1720. Файл с выдачей.
Для 10-го контига результаты оказались не менее интересными. Получилось 12 выравниваний, в основном длинной от 3 до 22 Kb. Суммарная длина выравнивания составила 80366 нуклеотидов. Доля контига, выравненная на геном, составила 80366/80969 * 100 = 99.26%. Участок хромосомы, куда произошло выравнивание контига — с 117349 по 198505 нуклеотиды. Число мисматчей — 13416, число гэпов — 1313. Ниже приведена сводная статистика по всем трём проанализированным контигам (см. Таблица 2.). Файл с выдачей.
| Номер контига | Длина контига (в нуклеотидах) | Процент покрытия хромосомы контигом | Число однонуклеотидных различий (mismatches) | Число гэпов | Доля контига, выравненная на хромосому (в процентах) | Участок хромосомы, соответствующий контигу |
|---|---|---|---|---|---|---|
| 6 | 107518 | 16 | 17193 | 1597 | 97.94 | 224334-330746 |
| 8 | 111992 | 17 | 17083 | 1720 | 98.40 | 460068-571665 |
| 10 | 80969 | 12 | 13416 | 1313 | 99.26 | 117349-198505 |