Парные выравнивания белков. Применение алгоритмов парных выравниваний к белку PDXK_BACSU
1. Сравнение матриц аминокислотных замен.
На сегодняшний день существует несколько типов матриц замен аминокислотных остатков.
Самая известная и широко используемая матрица весов– BLOSUM62. Она была разработана супругами Хеникофф(Steven Henikoff & Jorja Henikoff) в 1992 году на основе базы данных BLOCKS, в которой изначально были цитоплазматические белки. В базе BLOCKS находятся множественные выравнивания последовательностей белков. В блоках выравниваний могут встречаться практически идентичные последовательности, за счет которых частоты пар могут быть ошибочно «сдвинуты». Для того, чтобы этого избежать, последовательности на заданном % идентичности кластеризуют, а порог указывают в названии матрицы. Таким образом, 62 – порог кластеризации. Если процент идентичности последовательностей выше порога, то их считают одинаковыми.
Так как мембранные белки имеют свою специфику, встречаемость частот и замен аминокислот у них сильно сдвинута, применять к ним матрицу BLOSUM62 некорректно. Поэтому для таких белков в 2000 году была разработана матрица PHAT(PHAT: A Transmembrane-Specific Substitution Matrix) (Pauline Ng, Jorja Henikoff, Steven Henikoff).
Целью работы было построение матрицы BLOSUM62, исходя из таблицы частот встречаемости всех пар аминокислот. Построенную матрицу можно найти в таблице, там же приведены все расчеты.
Было проанализовано различие между значениями весов замен фенилаланина на другие аминокислоты и на саму себя для матриц BLOSUM62, классической и реконструированной, PHAT_T75_B73. Результаты приведены в таблице 1.
| BLOSUM62 | BLOSUM | PHAT_T75_B73 | ||
| G | Gly | -3 | -3 | -2 |
| P | Pro | -4 | -3 | -5 |
| C | Cys | -2 | -2 | 0 |
| S | Ser | -2 | -2 | -2 |
| T | Thr | -2 | -2 | -2 |
| N | Asn | -3 | -3 | -1 |
| Q | Gln | -3 | -2 | -2 |
| D | Asp | -3 | -4 | -5 |
| E | Glu | -3 | -3 | -5 |
| H | His | -1 | -1 | -2 |
| R | Arg | -3 | -3 | -7 |
| K | Lys | -3 | -3 | -7 |
| A | Ala | -2 | -2 | -1 |
| M | Met | 0 | 1 | 0 |
| I | Ile | 0 | 0 | 0 |
| L | Leu | 0 | 1 | 1 |
| W | Trp | 1 | -1 | 0 |
| F | Phe | 6 | 6 | 6 |
| Y | Tyr | 3 | 1 | 4 |
| V | Val | -1 | 3 | -1 |
Замена фенилаланина на самого себя
Фенилаланин является ароматической аминокислотой и участвует в гидрофобных стэкинг-взаимодействиях, играет важную роль в стабилизации белковых структур. Во всех матрицах значения веса замены для фенилаланина на самого себя равны 6.
Замена фенилаланина на аминокислоты, близкие по свойствам
Помимо фенилаланина ароматескими являются триптофан (W) и тирозин (Y). Значения в BLOSUM62 и PHAT практически не отличаются, но в реконструированной матрице они меньше на несколько единиц. Возможно, это связано с некоторыми различиями в исходных данных.
Замена фенилаланина на аминокислоты из других функциональных групп
Рассмотрим замену фенилаланина на положительно заряженные гидрофильные кислоты аргинин и лизин. В BLOSUM62 и в реконструированной BLOSUM вес замены равен -3, а в PHAT равен -7. Это значит, что в мембранных белках замена на положительные кислоты происходит намного реже.
Вес замен фенилаланина на глутаминовую и аспарагиновую кислоты, отрицательно заряженные гидрофильные аминокислоты, практически не отличается во всех трех матрицах, однако в PHAT он всё же меньше как минимум на 1, что, видимо, связано с большей консервативностью мембранных белков по сравнению с цитоплазматическими.
2. Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Существуют различные алгоритмы для парного выравнивания последовательностей. Одними из самых известных являются алгоритмы Нидлмана-Вунша и Смита-Ватермана.
Алгоритм Нидлмана-Вунша осуществляет глобальное парное выравнивание, то есть выравнивание полноразмерных последовательностей. Осуществляется с помощью команды needle пакета EMBOSS.
Алгоритм Смита-Ватермана создает локальные парные выравнивания, то есть ищет максимально сходные участки последовательностей.
Оба алгоритма основаны на методе динамического программирования.
Важным критерием выравнивания является наличие гэпов. Гэп – это пропуск аминокислоты в одной из последовательностей при выравнивании. За каждый гэп назначается определенный штраф. Важные параметры, которые используют алгоритмы: штраф за открывающий гэп (Gap opening penalty; параметр -gapopened) и штраф за продолжающий гэп, то есть гэп, стоящий рядом с другим (Gap extension penalty; параметр -gapextended). Штрафы по умолчанию равны 10 и 0.5 соответственно.
Результаты сравнения выравниваний последовательности белка pdxk_bacsu с искуственно созданной последовательностью из 20 аминокислот, полученных с помощью программ needle, water, а так же вручную приведены ниже. Выравнивания сравниваются по следующим параметрам:
- Длина выравнивания (length)
- Процент идентичности (identity) (вычисляется по формуле I=100*M/N,где N – длина выравнивания, M – число совпавших пар аминокислот)
- Процент сходства (similarity) (вычисляется по формуле I=100*M/N,где N – длина выравнивания, M – число совпавших пар аминокислот, причем совпадающими аминокислотами могут считаться кислоты со сходными свойствами бокового радикала.
- Вес выравнивания по матрице BLOSUM62 (score) (сумма значений для каждой пары аминокислот в выравнивании минус штрафы за гэпы).
Мутант 1. Вероятность изменения остатка (моделирующая "ошибку" ДНК-полимеразы) – 0.6; вероятность замены остатка в случае, если данная позиция будет изменена – 0.6.
Выравнивание вручную
- Length: 23;
- Identity: 18/23 (78%);
- Similarity: 18/23 (78%);
- Score: 54.
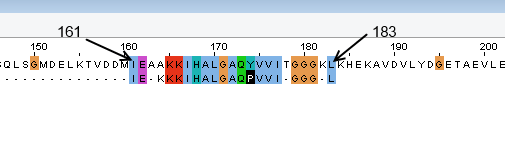
Выровненный фрагмент приведен на рисунке 1.
Needle
- Length: 16
- Identity: 16/271 ( 5.9%)
- Similarity: 16/271 ( 5.9%)
- Score: 58.0
Ниже представлен результат выравнивания - остатки исходной последовательности 161-182.
generations=1 1 ----------IE-KKKIHALGAQPVVIGGGL------------------- 20
|| .|||||||||.|||.||.
PDXK_BACSU 151 MDELKTVDDMIEAAKKIHALGAQYVVITGGGKLKHEKAVDVLYDGETAEV 200
Water
- Length: 16
- Identity: 14/16 (87.5%)
- Similarity: 14/16 (87.5%)
- Score: 64.0
Ниже представлен результат выравнивания - остатки исходной последовательности 165-180.
generations=1 4 KKIHALGAQPVVIGGG 19
|||||||||.|||.||
PDXK_BACSU 165 KKIHALGAQYVVITGG 180
Мутант 2. Вероятность изменения остатка (моделирующая "ошибку" ДНК-полимеразы) – 0.6; вероятность замены остатка в случае, если данная позиция будет изменена – 0.8.
Выравнивание вручную.
- Length: 21
- Identity 16/21 (76%)
- Similarity 17/21 (81%)
- Score: 64.0
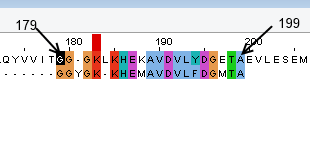
Выровненный фрагмент приведен на рисунке 2.
Needle
- Length: 271
- Identity: 14/271 ( 5.2%)
- Similarity: 15/271 ( 5.5%)
- Score: 66.0
Ниже представлен результат выравнивания - остатки исходной последовательности 179-198.
generations=1 1 ----------------------------GGYGKKHEMAVDVLFDGMTA-- 20
||...|||.|||||:||.||
PDXK_BACSU 151 MDELKTVDDMIEAAKKIHALGAQYVVITGGGKLKHEKAVDVLYDGETAEV 200
Water
- Length: 20
- Identity: 14/20 (70.0%)
- Similarity: 15/20 (75.0%)
- Score: 66.0
Ниже представлен результат выравнивания - остатки исходной последовательности 179-198.
generations=1 1 GGYGKKHEMAVDVLFDGMTA 20
||...|||.|||||:||.||
PDXK_BACSU 179 GGGKLKHEKAVDVLYDGETA 198
Мутант 3. Вероятность изменения остатка (моделирующая "ошибку" ДНК-полимеразы) – 0.4; вероятность замены остатка в случае, если данная позиция будет изменена – 0.8.
Выравнивание вручную
- Length: 20
- Identity: 17/20 (85%)
- Similarity: 18/20 (90%)
- Score: 70
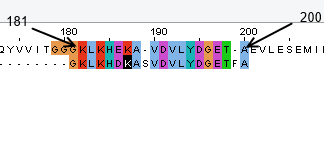
Выровненный фрагмент приведен на рисунке 3.
Needle
- Length: 16
- Identity: 16/271 ( 5.9%)
- Similarity: 16/271 ( 5.9%)
- Score: 58.0
Ниже представлен результат выравнивания - остатки исходной последовательности 184-201.
generations=1 1 -------------------------------EIRQEKAVDVYYFGYTAPK 19
:::.||||||.|.|.||..
PDXK_BACSU 151 MDELKTVDDMIEAAKKIHALGAQYVVITGGGKLKHEKAVDVLYDGETAEV 200
generations=1 20 L------------------------------------------------- 20
|
PDXK_BACSU 201 LESEMIDTPYTHGAGCTFSAAVTAELAKGAEVKEAIYAAKEFITAAIKES 250
Water
- Length: 16
- Identity: 14/16 (87.5%)
- Similarity: 14/16 (87.5%)
- Score: 64.0
Ниже представлен результат выравнивания - остатки исходной последовательности 182-201.
generations=1 1 EIRQEKAVDVYYFGYTAPKL 20
:::.||||||.|.|.||..|
PDXK_BACSU 182 KLKHEKAVDVLYDGETAEVL 201