Сборка генома de novo
Была осуществлена попытка сбора генома бактерии-эндосимбионта тлей Buchnera aphidicola str. Tuc7 на основании результатов секвенирования генома бактерии по технологии Illumina (SRR ID: SRR4240380).
Подготовка чтений
Чтения были скачаны с сайта ENA:
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/000/SRR4240380/SRR4240380.fastq.gz
Изначально при помощи TrimmomaticSE из чтений были удалены возможные остатки адаптеров:
TrimmomaticSE SRR4240380.fastq.gz SRR4240380.adapterless.fastq.gz
ILLUMINACLIP:adapters.fa:2:7:7
В результате были удалены 1.88% чтений.
Далее программа TrimmomaticSE была применена для удаления нуклеотидов с качеством прочтения ниже 20 с правых концов чтений и фильтрации чтений по длине (были оставлены только чтения, имеющие длины не менее 32 нуклеотидов):
TrimmomaticSE SRR4240380.adapterless.fastq.gz SRR4240380.trimmed.fastq.gz TRAILING:20
MINLEN:32
Для трех файлов fastq.gz был првоеден анализ при помощи fastqc:
fastqc [file]
После чего полученные результаты были объединены при помощи multiqc:
multiqc --fullnames *fastqc.zip
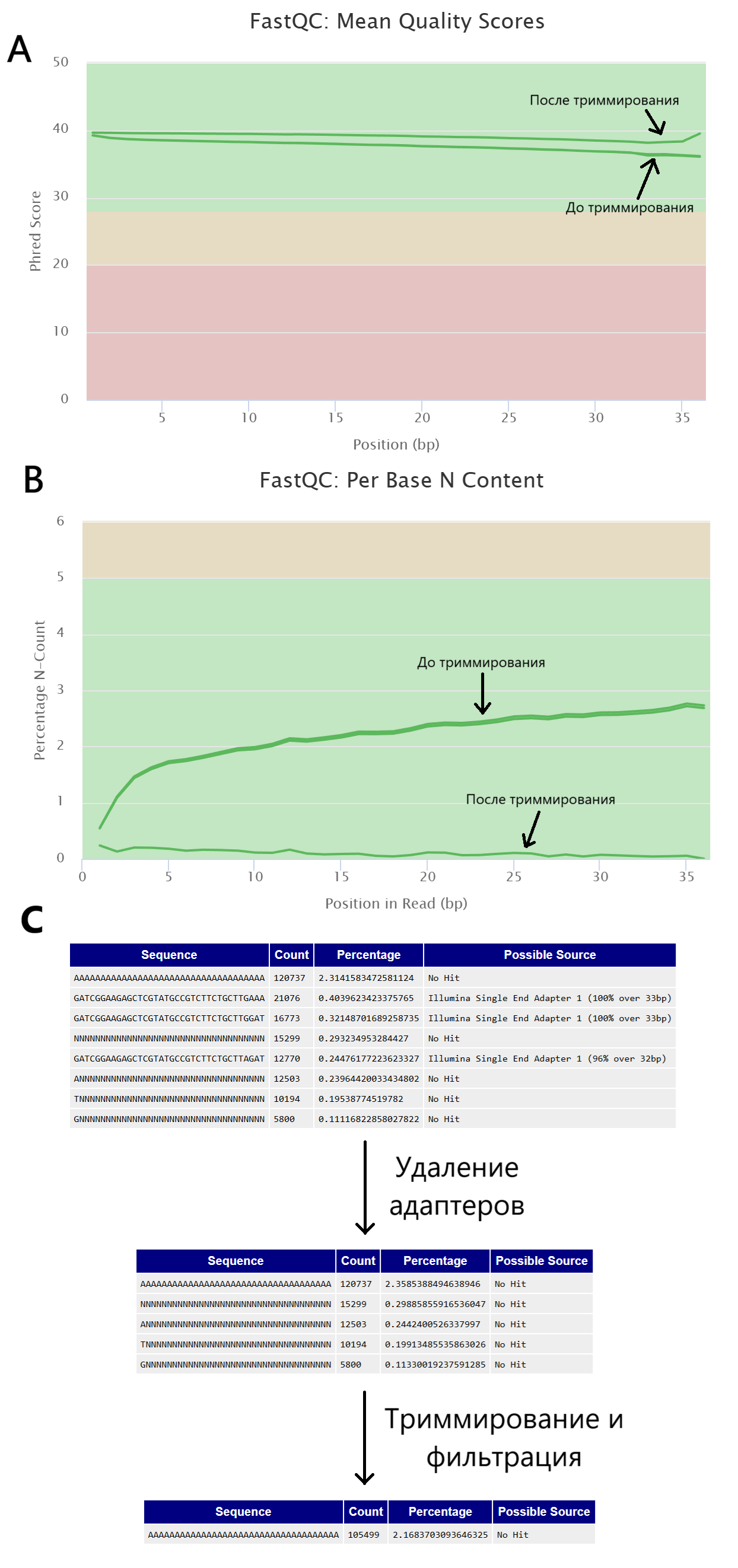
В результате из изначальных 5 217 318 чтений (размер файла - 112.4 Мб) после удаления адаптеров осталось 5 119 144 (98.1%, размер файла - 110.8 Мб), а после триммирования и фильтрации осталось 4 865 359 чтений (93.3% от изначальных, размер файла - 103.1 Мб).
Согласно отчету multiqc, качество последних позиций чтений возросло после триммирования и фильтрации, но не после удаления адаптеров (Рис. 1A), а также после работы TrimmomaticSE значительно уменьшилось количество неопределенных нуклеотидов в чтениях (N-Count, Рис. 1Б). Однако, среди оставшихся чтений все еще имелись сверхпредставленные поли-А последовательности (Рис. 1В).
Сборка при помощи velvet
Сначала при помощи velveth были подготовлены k-меры длиной 31 нуклеотид:
velveth kmers 31 -fastq.gz -short SRR4240380.trimmed.fastq.gz
Полученные k-меры были использованы для сборки при помощи velvetg:
velvetg kmers
Из файла contigs.fa была получена информация о собранных контигах и сохранена в вдие таблицы:
grep '>NODE' contigs.fa | tr -d '>' | tr '_' '\t' > contigs.tsv
Полученная таблица далее использовалась для анализа характеристик потенциальных контигов. Важно отметить, что значения длины, указанные в описаниях последовательностей показывают количество k-меров, из которых контиг был собран, поэтому действительная длина контига в нуклеотидах в данном случае будет на 30 больше (так как k-меры имели длину 31).В результате было получено:
Было получено 165 последовательностей
N50 для полученной сборки составило 12 072, L50 - 19
Самые длинные контиги имели длины 25 945, 23 880, 23 837 нуклеотидов и покрытия 27.4, 24.8, 25.7 соотвественно
Медиана покрытия контигов составила примерно 22.6
Из полученных контигов 2 имели покрытие более, чем в 5 раз большее, а 11 - более, чем в 5 раз меньшее, чем медианное
Контиги, имевшие очень высокое покрытие имели длину 2 136 и 964 нуклеотида (покрытия 126 и 130.5 соответственно). Эти два предполагаемых контига были выровнены на последовательность генома другого штамма бактерии - Buchnera aphidicola str. 5A (GenBank AC: CP001161). При использовании Megablast значимого сходства обнаружено не было, но при использовании blastn были получены результаты, представленные на Рисунке 2.
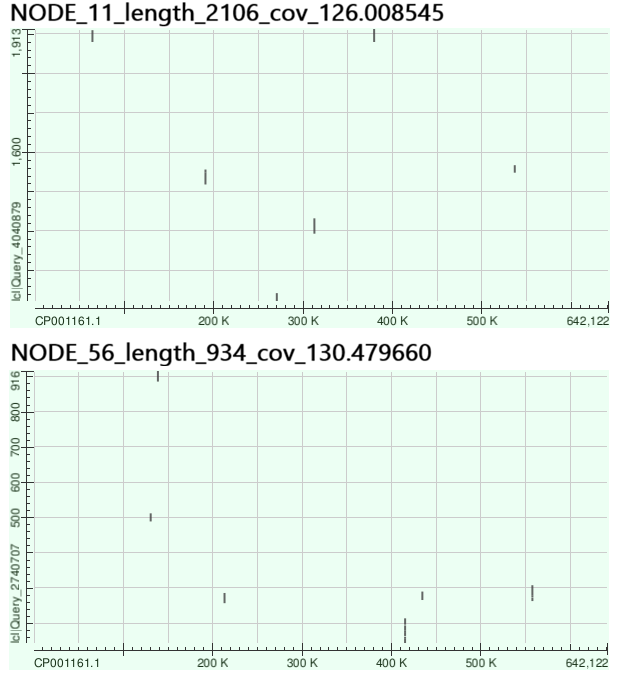
Как можно видеть, и первый, и второй пример выровнялись на геном в разных его участках, причем в выравнивания попали разные части последовательностей. Это говорит о том, что данные потенциальные контиги скорее всего не существуют как единые последовательности в геноме, а могли возникнуть, например, вследствие наличия в геноме бактерии повторов длиной более 31 нуклеотида (больше длины k-мера).
Контиги, имевшие низкое покрытие имели длину не более 122 нуклеотидов. Такие контиги скорее не должны учитываться при сборке генома, поскольку могут возникать, например, в результате контаминации.
Для трех самых крупных контигов были построены карты локального сходства (Рис. 3) с геномом Buchnera aphidicola str. 5A.
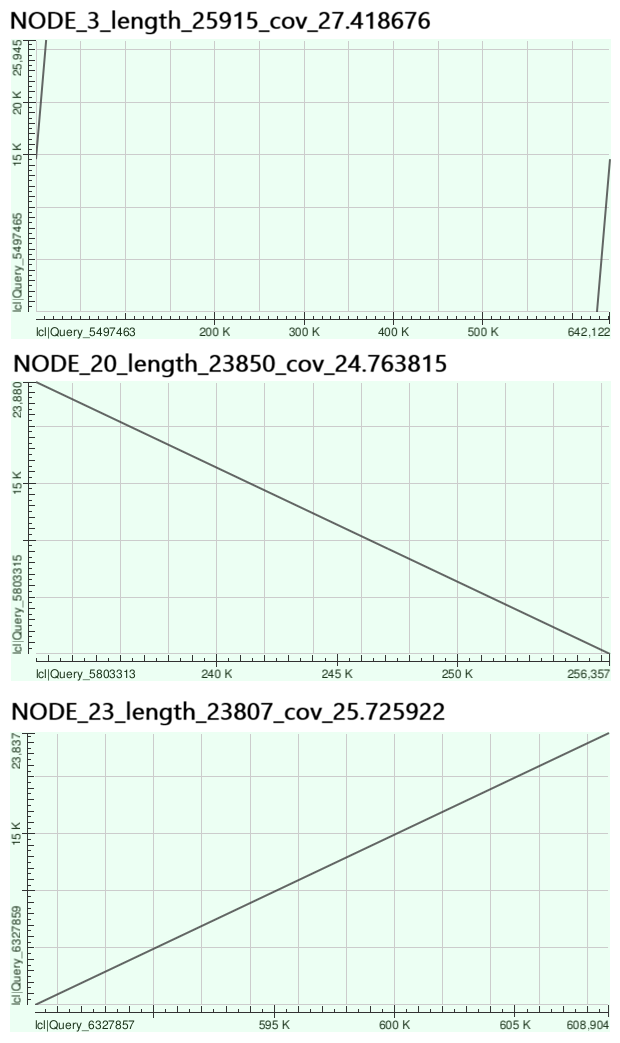
Самые длинные контиги: 3, 20 и 23, выровнялись на геном практически идеально (100% идентичности для контигов 3 и 23, только в вырванивании контига 20 имелось 2 гэпа), правда контиг 3 оказался разделен на 2 части точкой начала последовательности генома, а контиг 20 расположен на цепи, комплементарной той, чья последовательность представлена в GenBank. Таким образом, самые крупные контиги в данном случае действительно соотвествуют реально присутсвующим в геноме последовательностям.
Сборка с длиной слова 27
Для сборки также были использованы velveth и velvetg, но на этот раз k-меры имели длину 27 нуклеотидов:
velveth kmers27 27 -fastq.gz -short SRR4240380.trimmed.fastq.gz
velvetg kmers27
Из файла contigs.fa были получены данные о контигах в виде таблицы:
grep '>NODE' contigs.fa | tr -d '>' | tr '_' '\t' > contigs27.tsv
Были получены следующие результаты:
Было получено 357 последовательностей
N50 для полученной сборки составило 13 525, L50 - 14
Самые длинные контиги имели длины 59 823, 56 223, 25 975 нуклеотидов и покрытия 44.2, 45.4, 44.1 соотвественно
Очень большое число контигов имело низкое покрытие (220 менее 10 и 185 менее 5), поэтому медиана покрытия составила примерно 4.8
Среди контигов пять имели покрытие более 100
Как можно видеть, самые крупные контиги оказались длинее таковых при использовании длины слова 31, но при этом имелось огромное количество коротких контигов. Это может объясняться тем, что при меньшей длине слова сборка происходит менее специфично.
Было интересно проверить, являются ли самые длинные полученные контиги реальными последовательностями из генома. Поэтому при помощи megablast на геном бактерии Buchnera aphidicola str. 5A были выровнены контиги 19, 8 и 5 (Рис. 4).
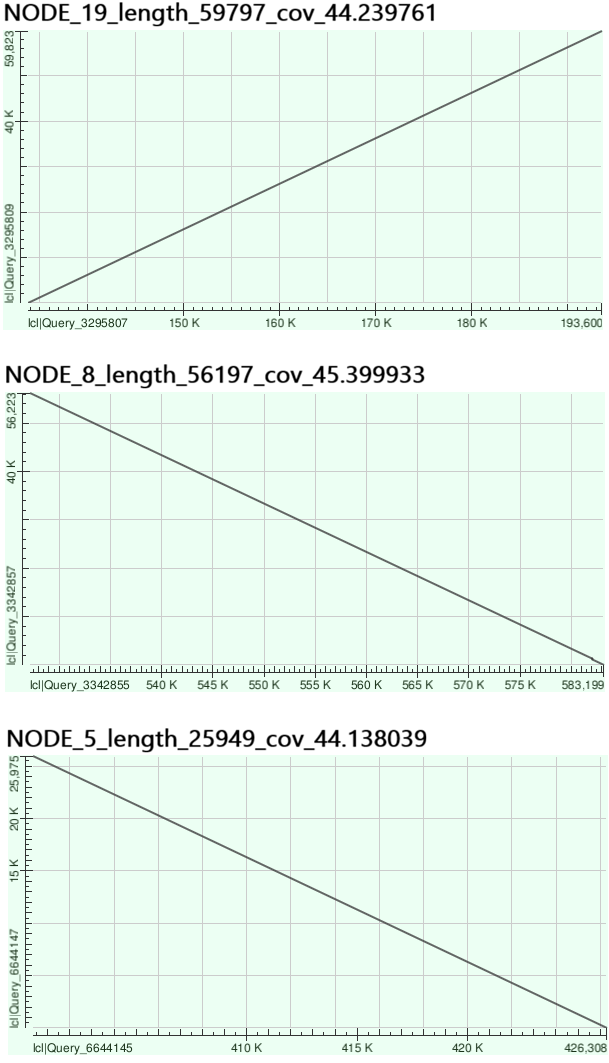
Как оказалось, данные контиги также соотвествуют существующим последовательностям в геноме, так как выровнялись почти идеально. Было интересно посмотреть на последовательности с аномально высоким покрытием, поэтому две крупнейшие из них (длинами 1 829 и 964) были выровнены на геном Buchnera aphidicola str. 5A (Рис. 5).
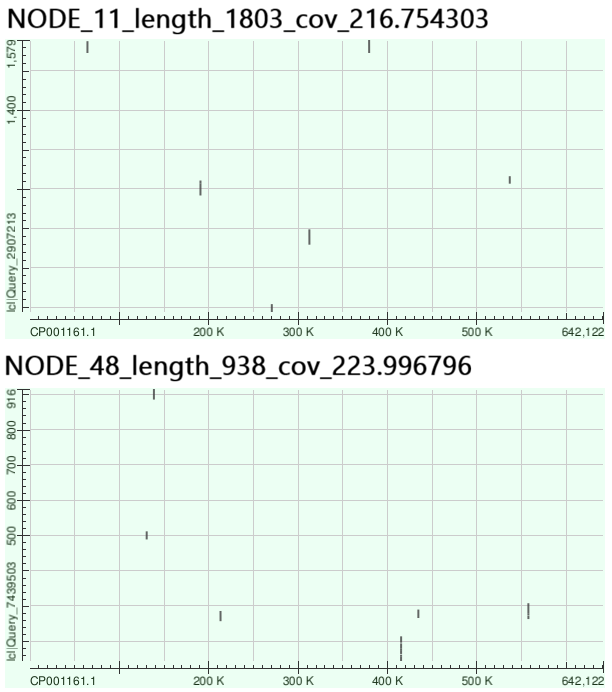
Можно видеть, что предполагаемые контиги 11 и 48 выровнялись на разные участки генома. Что интересно, последовательность 48 из сборки при длине слова 27 полностью совпала с последовательностью 56 из сборки с длиной слова 31 (которая также имела аномальное покрытие), аналогично последовательности 11 из обеих сборок почти полностью совпали. Это вполне объяснимо, если причиной аномального покрытия является наличие повторяющихся последовательностей.
Для того, чтобы понять при какой длине слова большая доля полученных в результате сборки последовательностей скорее всего присутсвует в геноме, на геном бактерии Buchnera aphidicola str. 5A при помощи Megablast были выровнены все полученные потенциальные контиги для длины слова 31 и длины слова 27. Результаты были загружены в формате csv и обработаны:
cat kmers31_megablast.csv | tr ',' '\t' | cut -f1,7,8 | tail -n +2 | tr '_' '\t' | cut
-f2,4,6-8 > megablast31.tsv
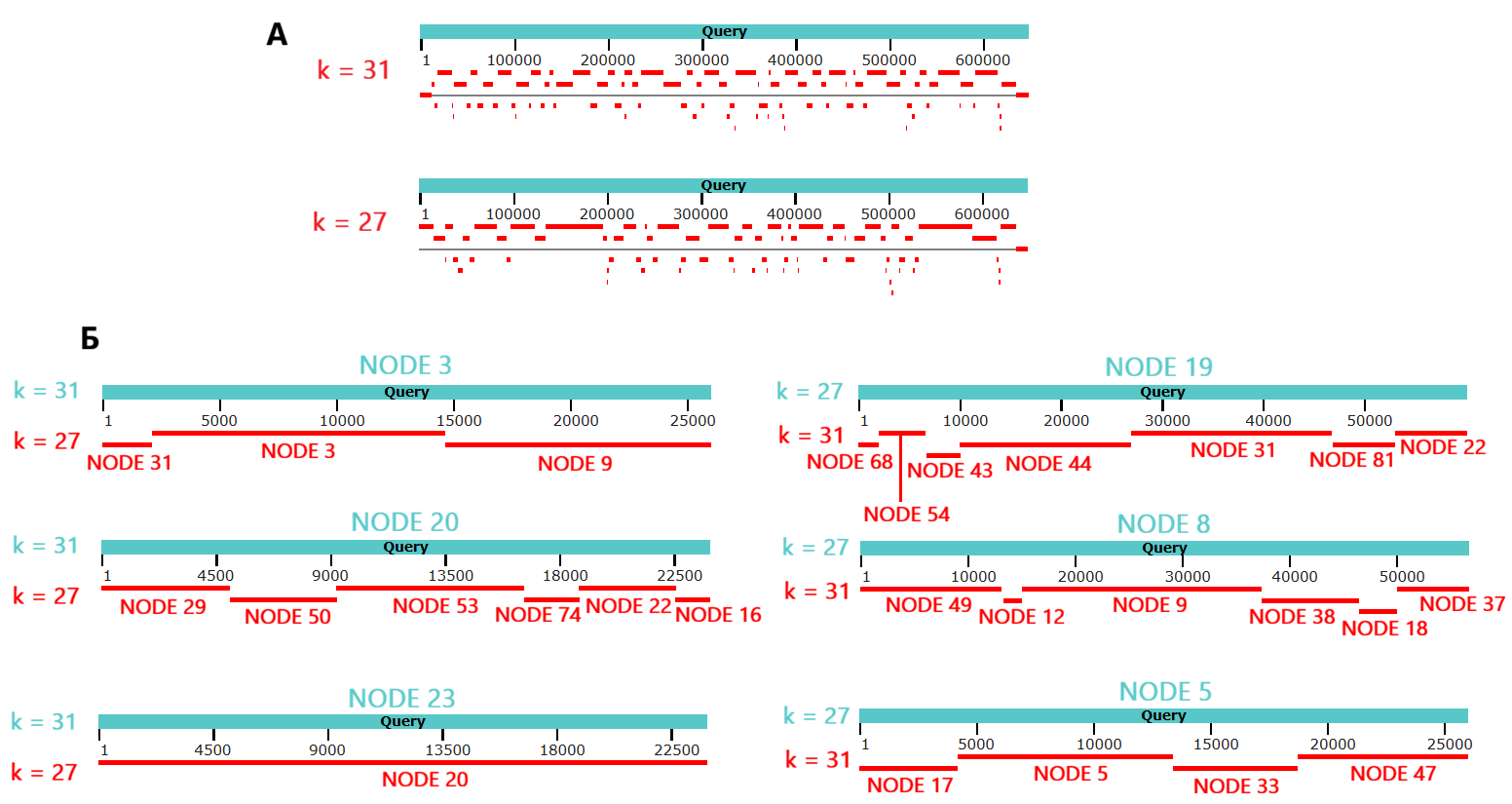
В результате для каждого предполагаемого контига из каждой сборки было определено, выровнялся ли он на геном при помощи Megablast. При длине слова 31 89 из 165 (53.9%) предполагаемых контигов были выровнены на геном, при длине слова 27 - 77 из 357 (21.6%), при этом суммарные длины выровненных контигов оказались примерно равны: 641 848 при длине слова 31 против 643 550 при длине слова 27. Также по результатам Megablast видно, что покрытым в обоих случаях оказался практически весь геном бактерии (Рис. 6А).
Также было выяснено (путем выравнивания с помощью Megablast каждого из трех длиннейших контигов одной сборки на все последовательности второй и наоборот), что каждый из трех самых крупных контигов из сборки с длиной слова 31 был собран в сборке с длиной слова 27 (но часто был поделен между несколькими контигами) и наоборот (Рис. 6Б).
В целом, наблюдаемые результаты можно объяснить тем, что при меньшей длине k-мера формируемый граф де Брейна оказывается более связным. С одной стороны, это приводит к тому, что в сборке появляются более длинные контиги (так как если два чтения имеют перекрытие длиной от 26 до 29 нуклеотидов, то они будут объединены при длине слова 27, но не будут при длине слова 31). С другой же стороны в более связном графе де Брёйна скорее всего будет большее количество трудноразрешимых для программы-сборщика узлов, что приведет либо к тому, что контиги, цельные при k = 31 окажутся разделены, либо к появлению фрагментов, не имеющих генетического смысла (отсюда огромное количество коротких последовательностей при k = 27).
Использование другого способа очистки
К чтениям с удаленными адаптерами был применен другой способ очистки - скользящим окнов ширины 5:
TrimmomaticSE SRR4240380.adapterless.fastq.gz SRR4240380.trimmed_alt.fastq.gz
SLIDINGWINDOW:5:20 MINLEN:32
Согласно отчету fastqc осталось 4 741 533 чтения (90.9% от изначального числа, размер файла - 98.2 Мб). Далее была проведена сборка при помощи velvet с длиной слова 31:
velveth kmers_alt 31 -fastq.gz -short SRR4240380.trimmed_alt.fastq.gz
velvetg kmers_alt
Из файла contigs.fa были получены данные о контигах в виде таблицы:
grep '>NODE' contigs.fa | tr -d '>' | tr '_' '\t' > contigs_alt.tsv
Описание результатов сборки:
Было получено 169 последовательностей
N50 для полученной сборки составило 12 072, L50 - 19
Самые длинные контиги имели длины 25 945, 23 880, 23 837 нуклеотидов и покрытия 27.4, 24.8, 25.7 соотвественно
Медиана покрытия контигов составила примерно 22.6
Из полученных контигов 3 имели покрытие более, чем в 5 раз большее, а 10 - более, чем в 5 раз меньшее, чем медианное
Как можно заметить, результаты получились практически идентичные сборке на основе чтений, очищенных не скользящим окном, это может быть связано в том числе с тем, что чтения исходно имели достаточно высокое качество, из-за чего результаты очистки почти не отличались.