 8 (926) 907 94 08
8 (926) 907 94 08
Всё на свете является чудом!
Реконструкция деревьев по нуклеотидным последовательностям. Анализ деревьев, содержащих паралоги.
Необходимо построить
филогенетическое дерево ранее отобранных
бактерий, но на этот раз используя
последовательности РНК малой субъединицы рибосомы (16S rRNA).
Для этого разделяем работу на несколько
последовательных этапов:
I. Найти последовательности 16S рибосомальной
РНК каждой из бактерий.
1 -
поиск записи
EMBL
для каждой бактерии
Для того чтобы найти запись EMBL, нам потребуется
AC (Accession code) каждой бактерии.
Это очень просто сделать при помощи сервиса на биоинформатическом
портале
ExPASy.
Вводя в окошко поиска мнемоники каждой бактерии получаем страницу с
информацией (запись EMBL).
2.1 - поиск в
соответствующей записи особенности "16S
rRNA"
Теперь банальным поиском по странице (Ctrl+F)
находим особенность rRNA в поле FT и выбираем ту особенность, где также
есть указание на "16S ribosomal RNA".
Для удобства скопировали необходимую информацию о каждой бактерии в
отдельный
файл.
Увы, в записи EBML, описывающей
полный геном бактерии STRPN, рРНК не
аннотированы.
Для того, чтобы всё же найти 16S рРНК в геноме этой бактерии,
воспользуемся программой
blastn на сервере
NCBI, которая, в частности, умеет быстро выравнивать две
последовательности (в данном случае полный геном бактерии STRPN
и 16S РНК близкородственной бактерии - STRP1).
Полный геном STRPN скачали из Европейского
Нуклеотидного Архива (ENA).
В результате, программа blastn выдала
выравнивание, которое дало нам возможность определить
координаты участка 16S rRNA для бактерии
STRPN. Полученные координаты тоже внесли в
файл
с информацией о каждой бактерии.
2.2 -
получение координат
начала и конца участка с этой особенностью
Из имеющего файла с информацией нам нужны только координаты участка.
Они записаны в ранее сделанном файле просто через две точки (пример:
741565..743071)
3 - вырезание
нужного участка из записи
EMBL
{kind=link}
seqret embl:"Accession code" -sask
Получили 8 файлов (так как всего 8 бактерий) вида bacsu_16S.fasta
II. Заполнить таблицу с информацией о бактериях.
Используя сделанный в пункте 2.1 файл, заполняем таблицу:
| Мнемоника | EMBL AC | Координаты 16S rRNA | Цепь |
| BACAN | AE017334 | 741565..743071 | прямая |
| BACSU | AL009126 | 9810..11364 | прямая |
| CLOB1 | CP000726 | 9282..10783 | прямая |
| CLOTE | AE015927 | 8715..10223 | комплементарная |
| STAA1 | AP009324 | 531922..533476 | прямая |
| STAES | AE015929 | 1598006..1599559 | комплементарная |
| STRP1 | AE004092 | 17170..18504 | прямая |
| STRPN | AE005672 | 15450..16784 | прямая |
III. Поместить все
последовательности в один файл, отредактировать их и выровнять.
Далее поместили все последовательности в один
файл,
при помощи команды cat:
cat bacsu_16S.fasta bacan_16S.fasta
clote_16S.fasta clob1_16S.fasta staa1_16S.fasta staes_16S.fasta
strp1_16S.fasta strpn_16S.fasta > all_bacteria_16S.fasta
Затем отредактировали последовательности, оставив только
мнемонику (BACSU, BACAN и т.д.).
После чего получили
выравнивание последовательностей, используя
программу muscle:
muscle -in all_bacteria_16S.fasta -out alignment_all_bacteria_16S.fasta
IV. Реконструировать
дерево, используя программу MEGA.
Полученное выравнивание открыли в программе MEGA
и реконструировали дерево методом "Maximum Likelihood" (Phylogeny
> Construct/Test Maximum Likelihood Tree...):
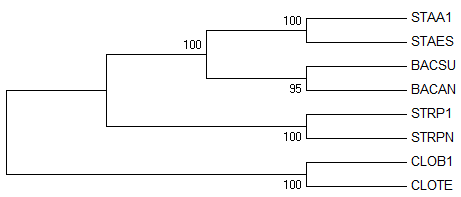 → реконструированное дерево
→ реконструированное дерево
V. Анализ дерева
в соответствии с правильным.
Ниже приведены для анализа реконструированное и правильное деревья:
→ реконструированное дерево
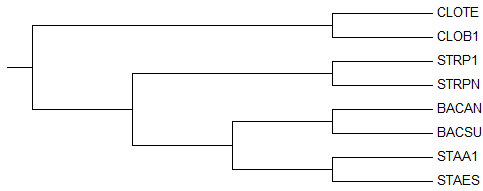 →
правильное дерево
→
правильное дерево
Полученное дерево полностью совпадает с правильным, поэтому, если всё
сделать аккуратно, то "реконструкция дерева по нуклеотидным
последовательностям" - это вполне рабочий и неплохой метод, хотя немного
муторный конечно в сравнении с методом "реконструкции дерева по белковым
последовательностям".
Но всё же, ошибок в дереве нет, поэтому, как говорится, победителей не
судят.
Необходимо найти в исследуемых бактериях достоверные гомологи белка
CLPX_BACSU. Для этого сначала получим
последовательность этого белка командой
seqret:
Затем по файлу proteo.fasta (там лежат записи банка UNIPROT, относящиеся к бактериям, перечисленным в таблице к первому заданию) создаем базу данных для поиска гомологов программой blastp:
formatdb -i proteo.fasta -p T -n base
После чего осуществляем поиск гомологов:
blastall -p blastp -d base -i clpx_bacsu.fasta -e 0.001 -o blastp.out
Теперь из полученного файла выбираем белки нужных нам бактерий.
Далее создаем лист-файл со списком идентификаторов ("адресов" последовательностей) отобранных белков, перед которыми стоит "sw:" (пример: sw:CLPX_BACSU).
Чтобы получить последовательности в fasta-формате, выполним команду seqret (знак "@" указывает программе seqret, что входной файл надо рассматривать как лист-файл, а не как файл с последовательностями):
seqret @myproteins_clpx_homologs.list myproteins_clpx_homologs.fasta
Полученные последовательности выровняли, используя программу muscle:
muscle -in myproteins_clpx_homologs.fasta -out alignment_myproteins_clpx_homologs.fasta
На выходе получили выравнивание (29-ти последовательностей белков), которое открыли через программу MEGA и реконструировали дерево методом "Maximum Likelihood" (картинка для "Bootstrap consensus tree"):
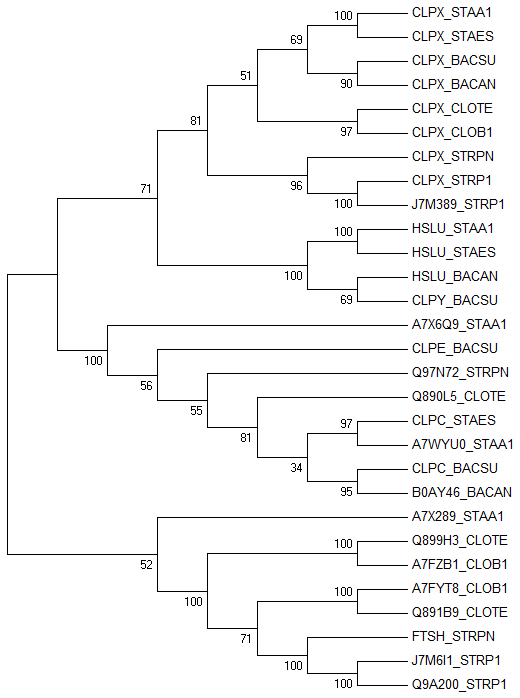
Внешне дерево производит впечатление вполне удачно реконструированного, но, в любом случае, считаем, что оно верное. Теперь определим ортологи и паралоги. Указание для их определения было дано в задании:
Два гомологичных белка будем называть ортологами, если они а) из разных организмов; б) разделение их общего предка на линии, ведущей к ним, произошло в результате видообразования.
Два гомологичных белка из одного организма будем называть паралогами.
Таким образом, приведем несколько пар ортологов и паралогов
(для удобства каждая пара выделена кружками (для ортологов) и ромбиками (для паралогов)):
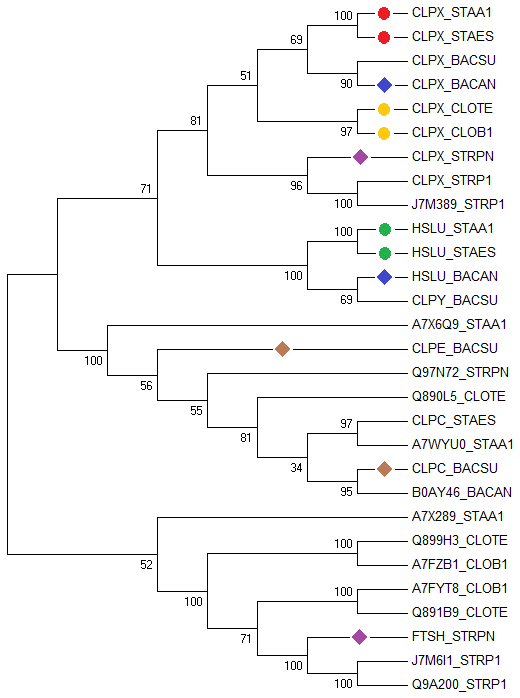
Ортологи
Паралоги
| Главная | Об авторе | Учебные семестры | Проекты автора | Друзья | Ссылки партнеров | Extra | Контакты |
Mneff © 2011-2013