1. Гомологи белка в Swiss - Prot
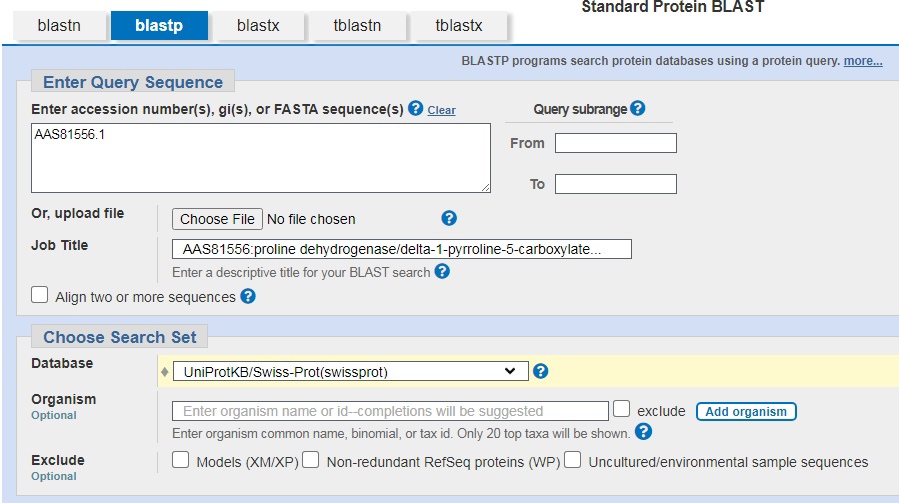
На рисунках 1 и 2 представлены параметры BLAST, используемые в исследовании.
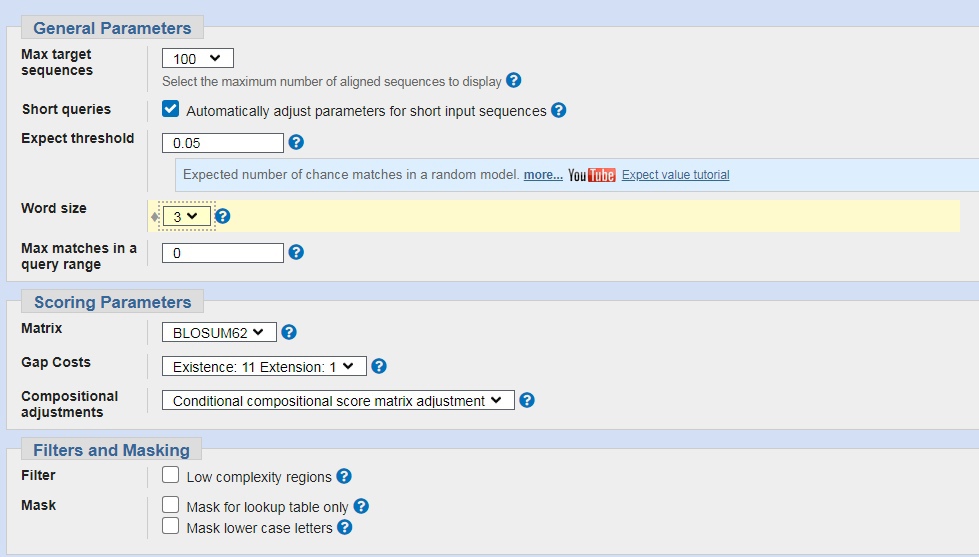
В самое большое окно был введён AC белка (хотя можно вводить и аминокислотную последовательность), в Database был выбран Swiss - Prot (как аннотированная база данных с проверенными последовательностями), алгоритм - blastp. В Algorithm parameters (дополнительных параметрах) максимальный размер выдачи последовательности на экран - 100, порог E - value (ожидаемое количество находок с таким же и лучшим весом выравнивания) составил 0.05, что позволило с большей вероятностью найти гомологичные белки. Длина слова была выбрана 3, это повысило чувствительность BLAST. Параметры выравнивания (матрица, штраф за гэпы) изменены не были.
Текстовую выдачу BLAST можно посмотреть по ссылке или другой ссылке.
Далее было произведено выравнивание в Jalview. Результаты работы можно посмотреть по ссылке. Кроме исходного белка организма Thermus thermophilus, по проценту идентичности (должен быть >20-25% для возможной гомологии), проценту покрытия и другим характеристикам были выбраны похожие белки Deinococcus radiodurans и несколько находок для Bacillus subtilis subsp. subtilis, Bacillus subtilis subsp. natto.
Комментарии:
Как видно из выравнивания, все последовательности можно назвать гомологичными, поскольку там довольно много консервативных участков, повторяющихся у всех белков: 1 столбец, 8 столбец, 12 столбец, а начиная с 187 столбца идёт область с высокой консервативностью, где очень много одинаковых аминокислот.
2. Гомологи зрелого вирусного белка, вырезанного из полипротеина в Swiss - Prot
При помощи UniProt качестве полипротеина для исследования был выбран Replicase polyprotein 1ab (ID: R1AB_CVMA5, AC: P0C6X9; O39225; O39226; P16342; P19750) вируса Murine coronavirus (strain A59) (MHV-A59) (Murine hepatitis virus). Один из зрелых белков в этом полипротеине - это Host translation inhibitor nsp1 (координата начала - 1 а.о., конца - 247 а.о.). Средствами EMBOSS был вырезан этот белок, последовательность можно посмотреть по ссылке.
Далее были проделаны те же действия, что и в пункте 1. При помощи BLAST с теми же параметрами были найдены возможные гомологичные белки для вируса. Текстовую выдачу BLAST можно увидеть по ссылке. На основании процента идентичности, процента покрытия и других параметров были выбраны 5 гомологичных белков (они принадлежат вирусам
Murine hepatitis virus strain A59, Murine hepatitis virus strain JHM, Murine hepatitis virus strain 2, Murine hepatitis virus strain defective JHM, также один гомологичный белок принадлежит исходному вирусу). Были скачаны
последовательности этих белков в fasta - формате, затем они были выровнены на kodomo при помощи команды mafft seqdumpfin.fasta > align_maftf.fasta
, отредактированы в Jalview, результаты работы можно скачать по ссылке.
Данные вирусные белки гомологичны, поскольку в выравнивании на протяжении всей последовательности (особенно в начале) наблюдается большое количество консервативных участков с одинаковыми или похожими аминокислотами. Также можно отметить, что белки неодинаковой длины, самые длинные - 1 (исследуемый), 4 и 6.
3. Исследование зависимости E-value от объёма банка
В следующем пункте в BLAST был проделан тот же запрос, но теперь поиск был ограничен только белками вирусов. Выдачу можно посмотреть по ссылке. Удивительно, но количество результатов не поменялось (осталось 23), однако можно заметить, что значение E - value для второго запроса в случае всех белков уменьшилось на несколько порядков (например, для самого первого результата E - value было 5e-172, а стало 2e-173, то есть оно уменьшилось примерно в 25 раз). Поскольку во втором запросе исследуемые организмы были ограничены только вирусами, уменьшился объём базы данных. Как известно, E - value прямо пропорционален этому объёму, поэтому значение E - value так же уменьшилось. Другие параметры не поменялись, следовательно, можно заключить, что база данных вирусных белков меньше примерно в 20 - 30 раз.
4. Сравнение интерфейсов BLAST
Сравнивались BLAST на сайте NCBI и Uniprot. Сначала поговорим про параметры, которые можно задать при поиске последовательностей. На NCBI параметров в принципе больше (можно выбрать с помощью from...to промежуток последовательности, который нужно искать; можно загружать файл, а не вводить только последовательность или ID; можно выбирать размер слова и т.д.). На Uniprot сложнее с выбором баз данных (их меньше, они представлены крупными таксонами), нельзя самостоятельно задать значение Expect threshold, Word size, меньше выбор матриц, труднее задать штрафы за гэпы и т.д. Однако при помощи Uniprot можно искать последовательности в базах данных Uniref и UniParc. Несмотря на то что в Uniprot меньше параметров и возможностей выбора, интерфейс кажется мне более простым и интуитивно понятным, чем на NCBI.
Стоит отметить, что поиск в Uniprot длится в принципе дольше, чем на NCBI.
Uniprot выдаёт каждый результат как бы отдельно, в то время NCBI выдаёт всё общим списком, где удобнее сравнивать Score, проценты покрытия, проценты идентичности и другие характеристики. На Uniprot меньше форматов, в которых можно скачать результаты работы. Однако на Uniprot вывод программы более наглядный, там есть цветная шкала, которая показывает Score, процент идентичности или E - value.
Таким образом, можно сказать, что BLAST на Uniprot проще будет использовать новичкам в этой сфере, потому что интерфейс там более интуитивный. Uniprot применим для локальных исследований небольшого количества последовательностей. NCBI можно использовать для изучения и скачивания большошо числа последовательней. Он более чувствительный, поэтому его можно использовать для исследований, где нужно задавать нестандартные параметры, которые, возможно, ориентированы под определённую последовательность.
5. Поиск "гомологов" бессмысленной последовательности
При помощи онлайн - ресурсов была сгенерирована случайная последовательность из 30 а.о. (YYSHTIGPATMSVLVGKFRSSIMPKWKLVA) и запрошена в BLAST с параметрами, указанными в пункте 1. Результаты можно посмотреть по ссылке.
Как можно заметить, ожидаемо результатов довольно много, потому что последовательность маленькая и может казаться гомологичной для разных участков больших протеомов. В таксономии можно заметить абсолютно разные и несвязанные группы организмов (бактерии, растения, животные, даже человек), что тоже неудивительно, поскольку последовательность бессмысленная, не несёт в себе каких - либо консервативных участков, которые могли бы быть характерны для определённой группы организмов. Банк данных был взять большой, поэтому и значение E - value тоже немаленькое (от 0.05 до >100). Также ожидаемое мал и вес последовательностей.
Сложнее было предполагать большие проценты идентичности и покрытия. Процент идентичности везде гораздо больше 20-25%, достаточных для гомологии двух белков. Возможно, это связано с тем, что была взята очень маленькая последовательность (всего 30 а.о.), поэтому велика была вероятность встречи такого сочетания букв в каких - либо организмах. Если бы последовательность была больше (хотя бы 200 - 300 аминокислотных остатков), то вероятность встречи похожего сочетния букв была бы меньше.