Домены и профили¶
Построение и проверка работы НММ-профиля семейства белков¶
Данный практикум нацелен на поиск наиболее оптимального порога для отнесения белка к группе с орпределенной доменной архитектурой с помощью ее HMM-профиля. Для этого в качестве исследуемого семейства были выбраны белки, содержащие домен MEDS (Pfam: PF14417), его функции плохо изучены. Данный домен удоволетворяет большинству необходимых критериев: всего последовательностей - 1803, средняя длина - 151.8, среднее покрытие - 43.37, средняя идентичность - 22% (это меньше 40, однако на результаты это, по все видимости, мало повлияло). Всего для данного домена в Pfam выявлено 126 архитектур с ним, один из которых включает 222 последовательности и включает два домена - MEDS в начале и STAS_2 (Pfam: PF13466) в конце (Рис.1.).
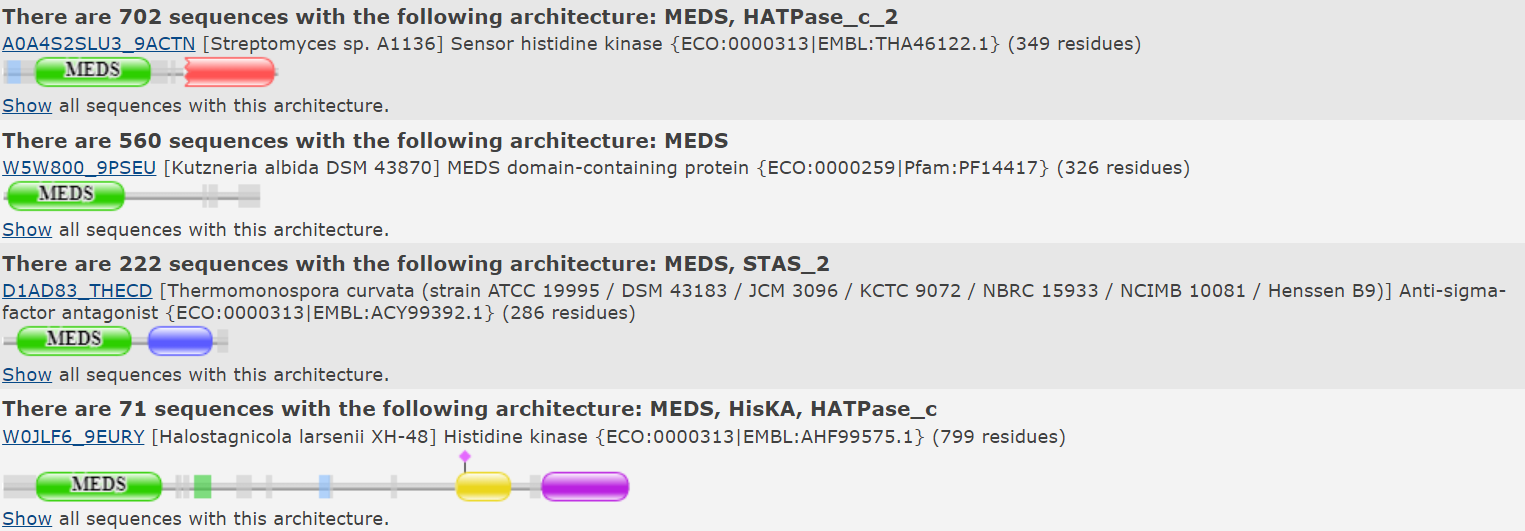
Число последовательностей в Seed домена составляет 110, в Full - 1803, в UniProt - 6629. Длина профиля HMM - 161.
Сперва из раздела Alignments был получен fasta-файл с полными последовательностями белков. С помощью скрипта, принимающего на вход файл с полными последовательностями и файл с идентификаторами последовательностей подходящей двухдоменной архитектуры, был получен fasta-файл с последовательностями, имеющими нужную архитектуру, на сонове которого с помощью программы MUSCLE было составлено выравнивание. В данном выравнивании были удалены неконсервативные области вначале и в конце, последовательности, у которых отсутсвуют протяженные части из консервативных участков и последовательности с 95%-ным сходством. Получившееся отредактированное выравнивание было подано на вход программе hmm2build, и были выполнны следующие команды:
hmm2build meds_arch_hmm.txt meds_aln_edited.fasta
hmm2calibrate meds_arch_hmm.txt
hmm2search --cpu 2 pgdh_c_arch1_hmm_mafft.txt pgdh_c_full.fasta > hmm_search_pgdh_c_mafft.txt
В результате был получен файл c HMM-профилем и файл c выдачей программы hmm2search, из которого была взята и переработана в формат CSV таблица с информацией о находках. С помощью другого скрипта, который принимает на вход файл с полными последовательностями, файл с идентификаторами последовательностей подходящей двухдоменной архитектуры и выше упомянутуютаблицу, были построены гистограмма распределения длин белков, график падения веса находок, график ROC кривой и график зависимости F1-меры от веса находки(Рис.2), а также отдельно была составлена таблица с расчетом значений для построения этих графиков (recall, precision, false positive rate, F1-score).

По полученным данным можно видеть, что длина подавляющего большинства белков лежит в пределах 240-300 а.о., а длина белков с исследуемой архитектурой - 280-320 а.о. (Рис.2.Sequence length distribution). На графике весов находок можно видеть, что для первых 500 последовательностей веса быстро падают примерно до -50, после чего начинают уменьшаться плавно примерно до -150, после чего снова резко падают (Рис.2.Score). Вероятно, такая картина получается в результате большого различия между последовательностями исследуемой доменой архитектуры. По построенной ROC кривой (Рис.2.ROC) видно, что полученный HMM-профиль с достаточной эффективностью может отличать последовательности с исследуемой доменной архитектурой от других последовательностей в семействе (ROC AUC = 0.98). Для наиболее эффективной классификации полученным HMM-профилем стоит выбрать порог равный 157.5, для которого F1-мера максимальна (Рис.2.F1-score). C подробной таблицей для построения F1-меры можно ознакомиться по следующей ссылке.