Работа с BLAST
Поиск гомологов белка в базе данных Swissprot
В этой части работы необходимо найти гомологи белка, который мы изучали в предыдущих практикумах. В нашем случае, это зеленый флюоресцирующий белок, полученный из медузы Aequorea victoria - GFP(P42212) Из Uniprot была скачана аминокислотная последовательность белка в формате fasta-файла. Поиск белков-гомологов осуществлялся в программе BLAST. При запуске этой программы были использованы следующие параметры:
Всего было выдано 20 предположительно гомологичных последовательностей. С текстовой выдачей BLAST можно ознакомиться по ссылке.
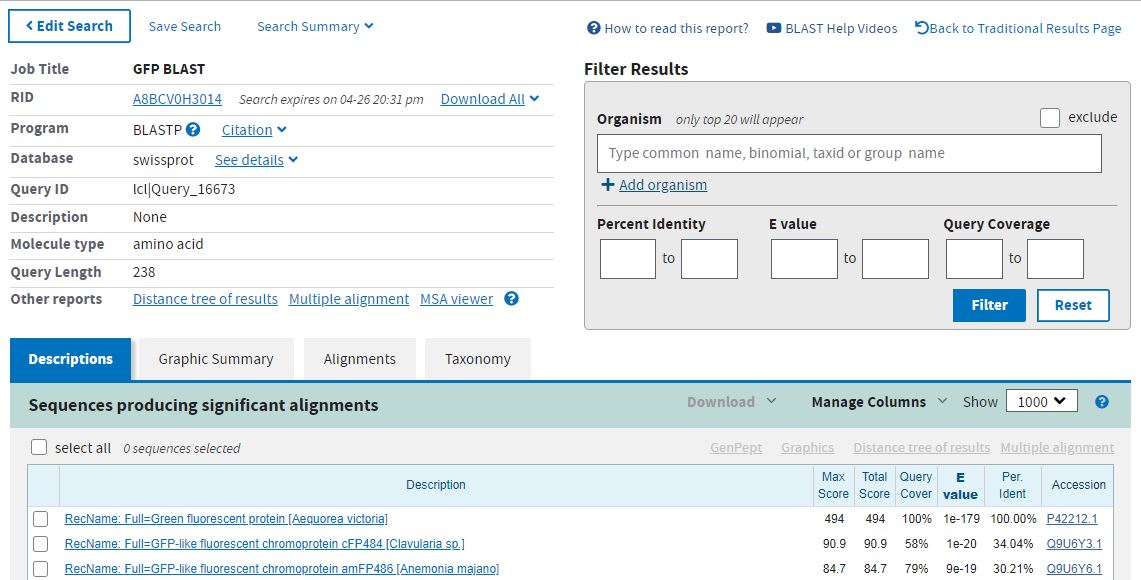
Интересно, что в список выдачи BLAST попали даже такие, на первый взгляд, отличные от искомого белки, как лейцин-тРНК лигаза и метилтрансфераза. Но у соответствующих
выравниваний очень большое значение E-value, поэтому можно не волноваться по этому поводу.
Для множественного выравнивания были выбраны 6 белков, все они обозначены как GFP-подобные хромопротеины, либо как флюоресцентные протеины.
BLAST предлагает скачать два варианта fasta-файла - один из них помечен как complete sequence, другой - как aligned sequence. Разница в этих файлах
заключается в том, что в complete sequence находятся полные последовательности белков, а в aligned - только выровненные участки, если процент покрытия не равен 100.
Файл с aligned sequence
можно посмотреть здесь, файл с
complete sequence - здесь
Выравнивание производилось с помощью программы muscle. Для выравнивания был выбран fasta-файл с полной последовательностью белков, чтобы лучше оценить гомологичность
[ссылка].
Далее выравнивание было визуализировано в
Jalview[ссылка].

Уже по выравниванию видно, что наименее похожим белком является как раз выбранный нами GFP. Это подтверждают также построенные на основе выравнивания деревья.
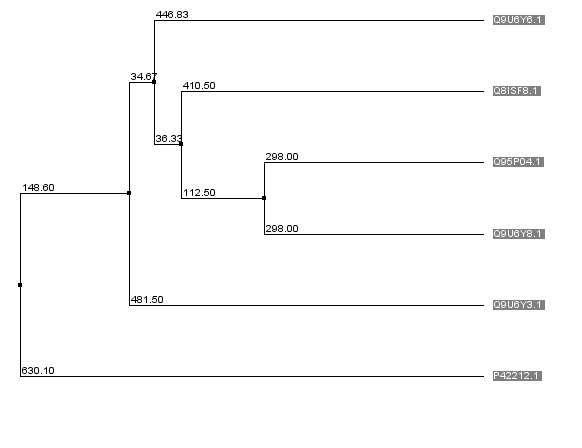
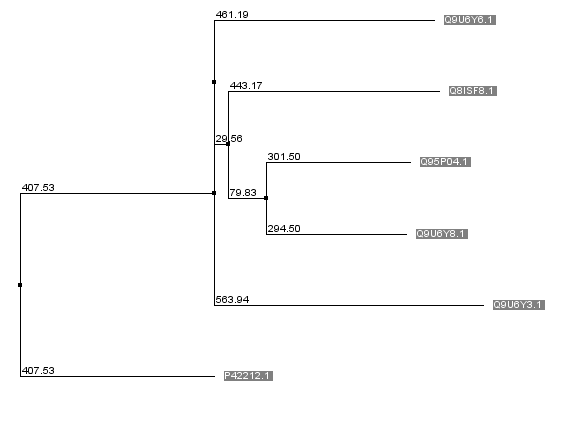
По деревьям также видно, что белок GFP(P42212) является аутгруппой в филогенетических деревьях. Однако похожие функции, родственные связи хозяев(все организмы-хозяева принадлежат к Типу Cnidaria), наличие нескольких консервативных участков и низкое значение E-value позволяет нам предположить, что это всё-таки гомологи, только, вероятно, они разошлись достаточно давно.
Поиск гомологов последовательности зрелого белка вируса MERS
В одном из прошлых практикумов нами был выделен зрелый белок из вируса MERS. Это была гуанин-N7-метилтрансфераза[ссылка на fasta-файл]. Она занимает позиции с 5909 по 6432 в полипептиде вируса.
Поиск гомологов осуществлялся в программе BLAST с теми же параметрами, что и в первой части работы. Результаты поиска содержали 31 полипротеин. Стоит отметить, что 22 результата имели E-value, равное 0, что
говорит о высокой достоверности находки, но ради интереса для выравнивания были взяты последовательности с E-value, отличным от нуля.
Для выравнивания было выбрано 5 белков различных вирусов, 3 из этих вирусов вызывают респираторные заболевания, как и MERS, а один вызывает свиную диарею, он был
выбран для того, чтобы посмотреть, будет ли его белок сильно отличаться от остальных:
Так как в задании написано, что необходимо удалить все буквы, которые не выровнены с какой-либо буквой исходного зрелого белка, будет лучше скачать fasta-файл с
aligned sequence. Результаты выравнивания можно скачать с проектом Jalview или посмотреть на рисунке 3.

По выравниванию можно видеть большое количество консервативных участков(6-9, 77-94, 104-122, 188-204, 270-284, 332-340, 378-389, 406-428, 473-484), малое количество
инделей, а учитывая очень маленькое значение E-value(степень приблизительно равна e-150), можно сказать, что эти белки являются гомологами.
Интересно, что гипотеза о большем отличии белка вируса, вызывающего свиную диарею, не подтвердилась. Он отличается от белка MERS не более, чем все остальные белки -
об этом говорит тот факт, что в консервативных участках его аминокислотная последовательность совпадает с последовательностью других 4 представителей.
Исследование зависимости E-value от объёма банка
В этом задании мы осуществили поиск того же белка вируса MERS, но в этот раз ограничили выборку только одним таксоном (Viruses). Затем мы сравнили E-value одних
и тех же последовательностей в запросе по всей базе данных[ссылка] и в пределах одного таксона[ссылка].
Достаточно интересно, что при поиске гомологов в пределах таксона Viruses количество находок увеличилось до 33 (при глобальном поиске их количество было равно 31).
Большая часть белков присутствует в обеих выдачах, но с разным значением E-value. Неудивительно, что значение E-value меняется, т.к. оно зависит от размера
выборки.
Рассмотрим изменение E-value нескольких находок (E0 - значение E-value при глобальном поиске, E1
- значение при поиске среди вирусов)
| Sequence ID | E0 | E1 | E1/E0 |
| P0C6Y2.1 | 2*10-173 | 8*10-175 | 0.04 |
| P0C6Y3.1 | 5*10-173 | 2*10-174 | 0.04 |
| P0C6X5.1 | 8*10-172 | 3*10-173 | 0.0375 |
| P0C6Y4.1 | 7*10-171 | 3*10-172 | 0.0428 |
E-value можно рассчитать по формуле Карлина, согласно которой его значение равно:
E-value = Kmn *e-λSK и λ - коэффициенты, зависящие от выбора матрицы выравнивания и штрафа за гэпы. В нашем поиске эти параметры были постоянными, поэтому эти
коэффициенты не изменялись. m и S не менялись, так как мы выравнивали одну и ту же последовательность. Получается, что в нашем случае E-value прямо
пропорционально размеру выборки. Из этого следует, что:
Получаем, что доля вирусных белков в базе данных Swissprot составляет примерно 4%
Сравнение интерфейсов BLAST
Для сравнения был выбран BLAST на сайте EBI. Ниже приведены скрины интерфейсов этих сайтов.
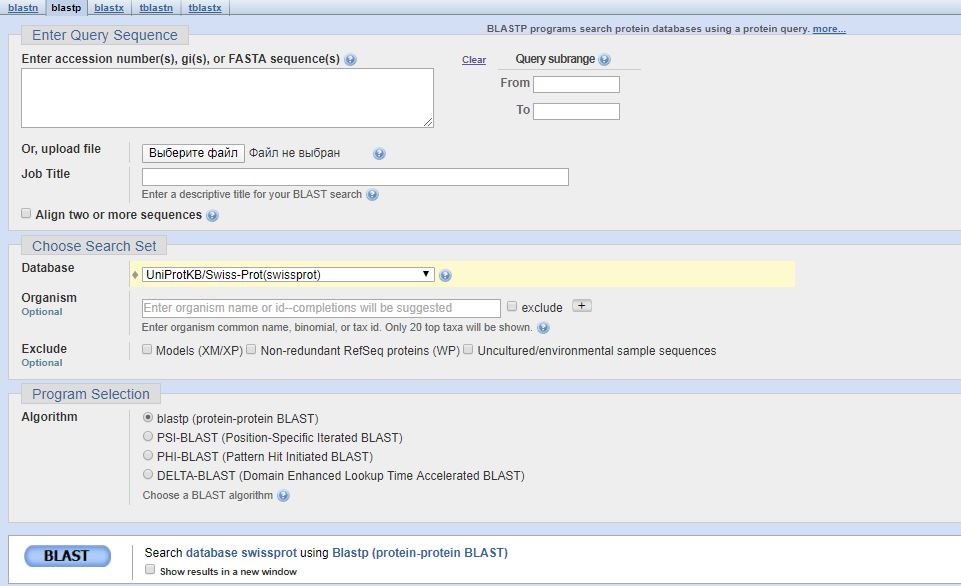
Сначала сравним поля ввода двух сайтов. В EBI намного более широкий выбор баз данных, в которых может вестись поиск гомологов. Там есть выбор между самыми
большими таксонами в UniProt KB, можно также выбрать кластеры в UniProt (в NCBI такая опция отсутствует), и наконец можно выбрать совершенно другие базы данных,
большую часть которых я не знаю.
В NCBI больше выбор алгоритмов поиска гомологов(blastp, PHI-BLAST, PSI-BLAST, DELTA-BLAST). В EBI представлены алгоритмы blastp и blastx.
В NCBI можно выбрать word size поиска из вариантов 2, 3 и 6. В EBI выбор шире - можно ввести любое число, но на больших числах или на
единице он вылетает или сильно виснет. В
любом случае, можно выбрать word size 4 или 5 и т.д.
В EBI есть опция Gapalign, которая определяет, допустимы ли гэпы в выравнивании или нет. На NCBI такой опции замечено не было. Зато в NCBI есть опция
Max matches in a query range, которая помогает найти гомологичные последовательности не только по одному участку последовательности, такой опции у EBI нет.
В остальном поля ввода совпадают, единственное, что в EBI можно выбрать формат вывода последовательностей-гомологов уже на стадии ввода (опция ALIGNMENT VIEWS)
и оставить почту, чтобы на неё пришло уведомление об окончании поиска
Теперь посмотрим на результаты поиска гомологов в BLAST на двух сайтах(для увеличения картинок нажмите на них).


В обоих сайтах мы получаем сводную таблицу результатов - в ней показываются вес выравнивания, процент идентичности и E-value. На сайте EBI также есть интересый раздел
Cross references and related information in, в котором содержатся ссылки, позволяющие больше узнать о предполагаемом белке-гомологе и о молекулах
или последовательностях, с ним связанных. Например, при нажатии на раздел Protein sequence в записи о GFP-подобном хромопротение
выдались его кластеры Uniref.
В NCBI можно отфильтровать находки по значению E-value или по проценту идентичности. Также в нём
есть возможность построения филогенетического дерева организмов-хозяев предположительно гомологичных белков. Можно задавать, например, метод построения
дерева или смотреть таксономическую принадлежность хозяев.
В EBI такой возможности не обнаружено, зато есть интересная вкладка под названием Functional Predictions, в которой, как я понял, указывается предположительно
функциональный домен. Вот такой файл
получен в результате поиска гомологов белка GFP на сайте EBI.
Подытоживая всё вышесказанное, можно сказать, что сайты достаточно сильно похожи, но в EBI лучше работать, если необходимо выбрать определенную базу данных для
поиска, т.к. их выбор там очень велик, либо, если необходимо более детально изучить предполагаемые гомологи.
{kind=link}
Поиск "гомологов" бессмысленной последовательности
Случайно сгенерированная последовательность была получена с помощью команды makeprotseq из пакета EMBOSS[
файл]. Сначала поиск гомологов производился на сайте NCBI
в БД Swissprot при таких же параметрах, как и в первой части работы. Результаты поиска гомологов представлены здесь.
По данным видно, что эти белки с очень малой вероятностью являются даже отдалёнными гомологами( например, фактор элонгации и субъединица АТФ-синтазы), на отсутствие
гомологичности указывает также значение E-value, близкое к пороговому значению и низкий процент покрытия и идентичности.
Ради интереса последовательность была уменьшена в 2 раза, чтобы проверить, найдётся ли большее количество гомологов, если последовательность будет короче.
Это было бы логично, ведь если мы сравниваем короткий кусок с последовательностями, больше вероятность, что он совпадёт.
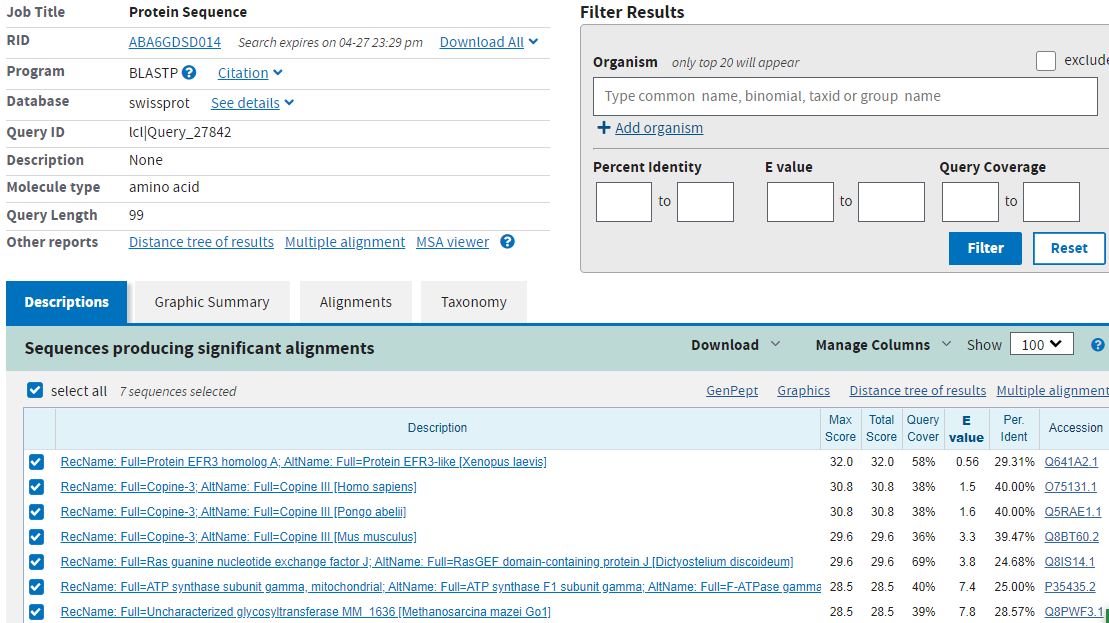
Результаты поиска можно посмотреть
здесь.
Интересно, что E-value у субъединицы АТФ-синтазы с уменьшением последовательности увеличился, а вот у гомолога белка ERF3 он, наоборот, уменьшился.