Нуклеотидный BLAST
Определение функции и таксономии нуклеотидной последовательности
В предыдущем практикуме на основании данных
о секвенировании прямой и обратной цепочек ДНК мы получили консенсусную
последовательность. Теперь нам необходимо определить функцию полученной последовательности и предположить, какому организму она принадлежит.
Для выполнения данного задания мы
воспользовались сервисом BLAST. Перед поиском гомологичных последовательностей нам необходимо было задать параметры этого самого поиска.
Мы не знаем, является ли наша последовательность консервативной, может быть, у неё очень мало или совсем нет очень похожих на неё гомологов, поэтому вариант поиска
с помощью megablast не подходит - слишком большая длина слов, много последовательностей может быть пропущено из-за этого (недостаточно чувствительный).
Discontiguous megablast видится ненужным для поиска, так как есть более простой вариант, а именно blastn, в котором можно взять меньшую длину слова.

После того, как мы выбрали конкретный алгоритм для поиска, мы настроили некоторые параметры алгоритма. Так, мы выбрали наименьшую длину слова, чтобы найти как можно больше гомологичных последовательностей, параметр Expected threshold мы оставили по умолчанию - 0.05, потому что мы хотим найти последовательности, которые с наибольшей вероятностью будут гомологичными.. Все остальные параметры были установлены по умолчанию. В результате мы получили вот такой список последовательностей (скачать выдачу BLAST можно здесь).
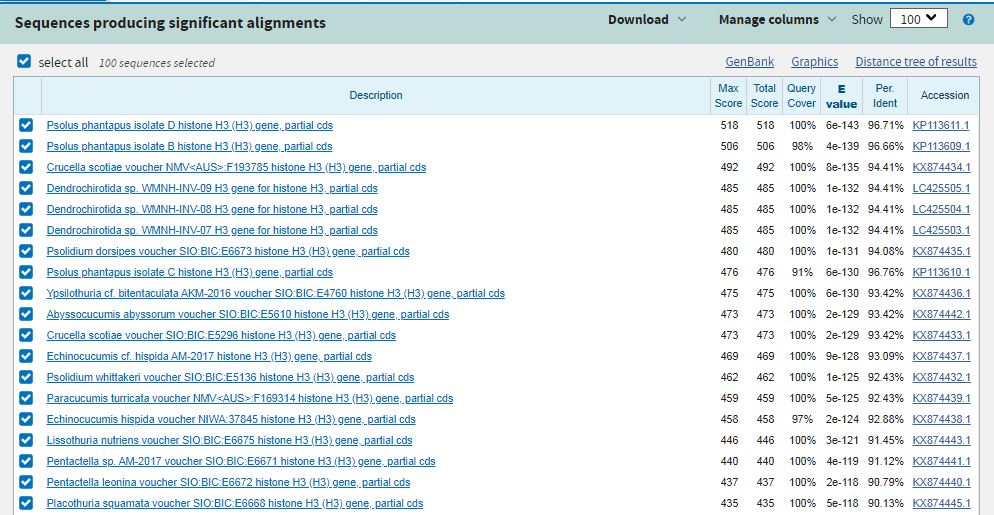
Все найденные последовательности кодируют гистон H3 (важно отметить, что, судя по всему, наша последовательность
является лишь частью гена, кодирующего этот белок - на это указывает подпись partial во всех находках).
Находки принадлежат к разным организмам,
в частности, практически идентичная нуклеотидная последовательность встречается у вида Psolus Phantapus, а также у Abyssocucumis
Abyssorum и у Crucella scotiae. Понятно, почему так произошло - гистоны являются очень консервативными белками, соотвественно, и
кодирующие их нуклеотидные последовательности тоже являются достаточно консервативными.
Для утверждения уровня таксономии мы скачали выровненные последовательности (опция download -> aligned sequences) и визуализировали это выравнивание в JalView.
Результат можно видеть на изображении ниже.

В выравнивании мы посчитали число замен на данном участке - мы пренебрегали заменой одного нуклеотида в одной последовательности, поэтому за замену считали тот
столбец, в котором отличающиеся нуклеотиды встречались хотя бы в двух последовательностях. То есть, если мы видим, что у всех в 50 позиции стоит аденин, а у одного
организма - гуанин, мы эту позицию не считаем за место замены нуклеотида, если же гуанин как минимум у двух последовательностей, то эта позиция удовлетворяет
нашим критериям.
Было посчитано, что замены происходят примерно в 26 местах при общей длине фрагмента 304, что составляет примерно 9% от всей длины участка.
Здесь важно помнить, что у нас лишь часть гена, а не весь ген, но можно предположить, что количество замен на всем гене
будет составлять такую же часть от общей длины последовательности.
По итогу, на основании имеющихся у нас данных можно заключить, что имеющаяся у нас последовательность кодирует гистон H3 и, вероятнее
всего, принадлежит организму из отряда
Dendrochirotida .
Поиск генов белков в неаннотированной нуклеотидной последовательности
Для этого задания был взят контиг из организма (крапчатая утка), геном которого мы анализировали
в прошлом практикуме.
Длина взятого контига составляет примерно 15 000 нуклеотидов,
скачать его можно по ссылке. Для того, чтобы найти, есть ли
кодирующие белок гены в этом контиге, и если есть, то какой белок они кодируют, мы воспользовались программой blastx. На первом запуске мы оставили все настройки
по умолчанию, потому что не было известно, что мы на выходе получим.В качестве базы данных брали Non-reduntant protein sequences, чтобы на выходе
получить как можно больше похожих белковых последовательностей.
В результате получили вот такую
выдачу. По результатам можно предположить, что в контиге
присутствует ген, кодирующий альфа-4 субъединицу ламинина. Об этом говорит то, что почти все последовательности в выдаче представляют собой именно этот белок,
а также то, что организмы, белки которых выровнялись с контигом, являются родственными крапчатой утке, из которой и был взят этот контиг (по
крайней мере, они принадлежат к тому же классу). Так, например, в выдаче
встретилась американская савка (Oxyura jamaicensis), хохлатая кариама (Cariama cristata) и пингвин Адели (Pygoscelis adeliae).
Учитывая, что длина альфа-4 субъединицы ламинина составляет примерно 1800 аминокислотных остатков (данные взяты с Uniprot), а в выравнивании BLAST всего 70 аминокислот,
можно сказать, что в контиг попала только маленькая часть гена, кодирующего LAMA4 protein. Можно попробовать найти контиг с недостающей частью гена, но это сложно,
так как некоторые контиги содержат по 500 000 нуклеотидов, BLAST их просто не принимает.
Для того, чтобы немного разбавить текст, вставлю сюда фото савки, кариамы и пингвина Адели.



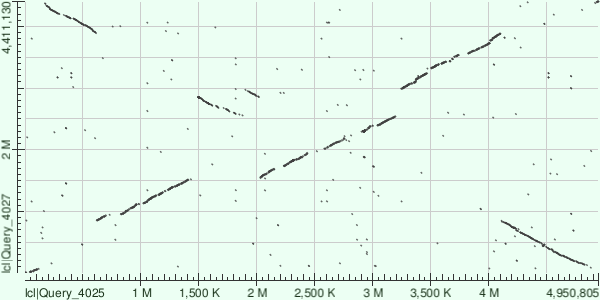
Интепретация карты локального сходства гомологичных хромосом двух бактерий
Для сравнения мы выбрали две бактерии из одного рода - это бактерия Mycobacterium tuberculosis(геном в формате fasta),
возбудитель туберкулеза, и Mycobacterium avium(геном в fasta-формате), возбудитель
микобактериоза и вообще деструкции легочной ткани у людей с иммуносупрессией.
Геном выбранных бактерий составляет примерно 4.5-5 мегабаз, но карта получилась неплохая, и было решено её оставить в отчёте. По горизонтали отложен геном Mycobacterium avium
(NZ_AP012555.1),
по вертикали - Mycobacterium tuberculosis (NC_000962.3)
На этой карте можно заметить крупную инверсию на участке 4,100K-600K у M.avium 800K - 3900K у M.tuberculosis. Также видно, что несколько средних по длине
участков (600К - 1400К по горизонтальной оси и 3,200К - 4.100К по горизонтальной оси) расположены выше основной диагонали, может быть, это произошло из-за разницы в длине геномов
сравниваемых бактерий, может быть, это транслокации (хотя, скорее нет).
На участке 750К-850К M.avium виден индель - ему нет соответствия в геноме M.tuberculosis (либо выравнивания на этом участке были отфильтрованы из-за низкого значения E-value).
Тут либо произошла вставка у M.avium, либо делеция у M.tuberculosis.
Также заметны неконсервативные участки в выравнивании, в частности, они видны на 2,150К-2,200К M.avium - 1,8-1,9М M.tuberculosis, и на участке 2,450-2,600К M.avium -
1,950К-2,050К M.tuberculosis.
Ещё заметна предполагаемая инверсия на участке 1,500К-1,900К M.avium - 2,900-2,600К M.tuberculosis.