0. Создание рабочей директории.
Создадим рабочую директорию /nfs/srv/databases/ngs/yuliafan и скопируем туда файлы с ридами (chr20.fastq) и хромосомой (chr20.fasta).
1. Анализ качества чтений.
Контроль качества чтений с помощью программы FastQC.
2. Очистка чтений.
Очистка чтений с помощью программы Trimmomatic. Отрезает с конца каждого чтения нуклеотиды с качеством ниже 20 (TRAILING:20)
и оставляет только чтения длиной не меньше 50 нуклеотидов (MINLEN:50).
Еще раз сделаем контроль качества чтений с помощью программы FastQC.
| Анализ качества чтений | fastqc chr20.fastq |
| Очистка чтений | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr20.fastq chr20_trim.fastq TRAILING:20 MINLEN:50 |
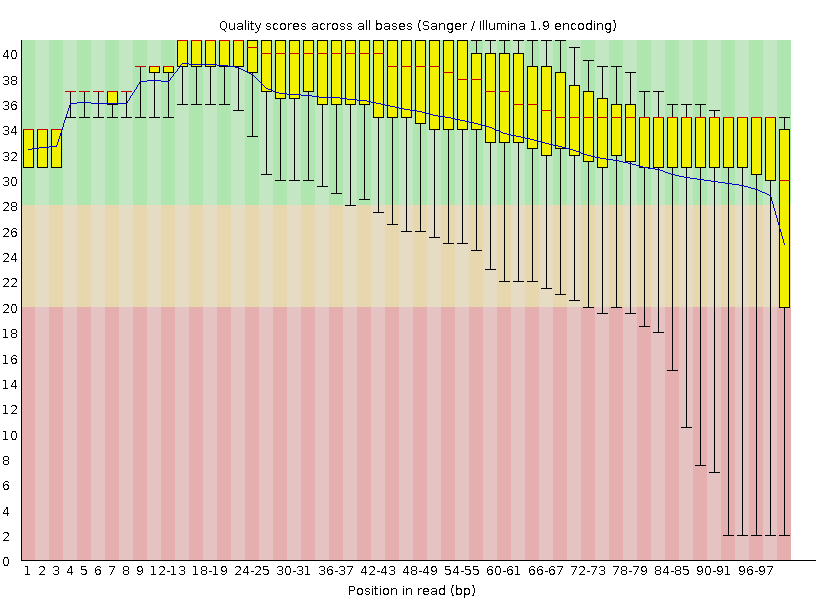
Рис 1. FastQC "Per base quality" до чистки
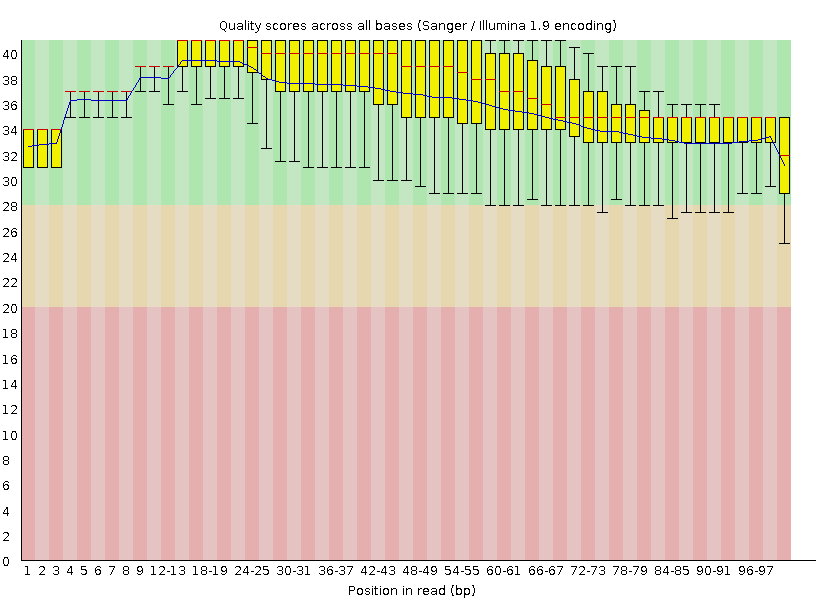
Рис 2. FastQC "Per base quality" после чистки
Число последовательностей после обработки уменьшилось с 4661 до 4472.
Остались чтения с качеством выше 20 и длиной не меньше 50 нуклеотидов.
3. Картирование чтений и 4. Анализ выравнивания
число чтений, картированных на хромосому: 4456
число чтений, не картированных на хромосому: 17
| hisat2-build | Индексирует референсную последовательность | hisat2-build chr20.fasta chr20 |
| hisat2 с параметрами --no-spliced-alignment и --no-softclip | Строит выравнивание прочтений и референса в формате .sam. | hisat2 -x chr20 chr20_trim.fastq --no-spliced-alignment --no-softclip > align.sam |
| samtools view | Переводит выравнивание чтений с референсом в бинарный формат .bam | samtools view align.sam -b > align.bam |
| samtools sort | Сортирует выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения | samtools sort align.bam -o sort.bam -T align.txt |
| samtools index | Индексирует отсортированный .bam файл | samtools index sort.bam |
| samtools idxstats | Выдаёт статистику в индексном файле, соответствующем входному файлу. | samtools idxstats sort.bam > idxstats |
5. Поиск SNP и инделей и 6. Аннотация SNP.
| samtools mpileup | Создаёт файл с полиморфизмами в формате .bcf | samtools mpileup -f chr20.fasta -g sort.bam -o snp.bcf |
| bcftools call | Создаёт файл со списком отличий между референсом и чтениями в формате .vcf | bcftools call -cv snp.bcf -o snp.vcf |
| Скрипт: convert2annovar.pl | Скрипт создаёт файл для последующей аннотации | perl /nfs/srv/databases/annovar/convert2annovar.pl.old -format vcf4 snp.vcf > snp.avinput |
| Скрипт: annotate_variation.pl | Аннотация snp на основе refgene | annotate_variation.pl -out refgene -build hg19 -dbtype refGene snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| Скрипт: annotate_variation.pl | Аннотация snp на основе dbsnp | annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| Скрипт: annotate_variation.p | Аннотация snp на основе 1000 genomes | annotate_variation.pl -filter -out 1000g -build hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| Скрипт: annotate_variation.p | Аннотация snp на основе GWAS | annotate_variation.pl -regionanno -out gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| Скрипт: annotate_variation.p | Аннотация snp на основе Clinvar | annotate_variation.pl -filter -out clinvar -build hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| Координата | Тип | Было | Стало | Качество | Покрытие |
| 33974207 | Замена | A | G | 207.009 | 39 |
| 34025756 | Замена | A | G | 221.999 | 41 |
| 56179934 | Замена | C | T | 101.008 | 20 |
В ходе работы я получила 42 snp, индели отсутствуют.
Качество и покрытие у большинства найденных полиморфизмов плохое.

База данных refseq в annovar делит snp на exonic, intergenic, intronic, upstream, UTR5.
У меня в эти группы попало: Exonic - 8, intergenic -3, intronic - 26, upstream -2, UTR5 – 1.
Мои snp попали в гены GDF5, SPATA2, ZBP1 (только полиморфизмы, попавшие в экзоны).
Snp привели к таким заменам: несинонимичные (4), синонимичные (4). С заменами можно ознакомиться в сводной таблице на вкладке refgene_exonic.
31 snp имеет rs.
Частота найденных snp:

Клиническая аннотация snp:
По данным GWAS найдено 3 snp, которые отвечают за наличие какой-либо черты или болезни - Height, Psoriasis, Hippocampal atrophy.
По данным Clinvar найден 1 snp, который влияет на артроз бедра.
Сводная таблица, в которую входят все snp и их характеристики: summary.xlsx
© Наумова Юлия, 2018