Задание 2.Сравните выравнивания одних и тех же последовательностей тремя разными программами
Для выполнения данного задания была использована программа Ксении Джолос.ссылка
Были выбраны следующие программы для сравнения - MAFFT, MUSCLE, ClustalO и T-Coffee. Причем выравнивание программы MAFFT было выбрано как референсное.
Белки были взяты из практикума 9(п.5) - DNAJ_YERPA, DNAJ_LEPBA, DNAJ_BACLD, DNAJ_BRUMB, DNAJ_EXIS2.
Ссылки на проект в Jalview:
Сравнение MAFFT и MUSCLE
ali_1.len: 404
ali_2.len: 398
%ali.col1: 78.96%
%ali.col2: 80.15%
| Блоки | Длина |
|---|---|
| (2-38)=(2-38) | 37 | (40-73)=(40-73) | 34 | (76-77)=(76-77) | 2 | (81-82)=(81-82) | 2 |
| (136-196)=(130-190) | 61 |
| (201-313)=(195-307) | 113 | (315-373)=(309-367) | 59 | (378-382)=(372-376) | 5 | (396-400)=(390-394) | 5 |
Одинаково выровненныe колонки, не входящиe в блоки отсутсвуют
Сравнение MAFFT и ClustalO
ali_1.len: 404
ali_2.len: 393
%ali.col1: 75.25%
%ali.col2: 77.35%
| Блоки | Длина |
|---|---|
| (2-38)=(2-38) | 37 |
| (41-74)=(41-74) | 34 |
| (134-196)=(124-186) | 63 |
| (202-313)=(192-303) | 112 |
| (316-372)=(306-362) | 57 |
Одинаково выровненныe колонки, не входящиe в блоки отсутсвуют
Сравнение MAFFT и T-Coffee
ali_1.len: 404
ali_2.len: 395
%ali.col1: 78.22%
%ali.col2: 80.00%
| Блоки | Длина |
|---|---|
| (3-38)=(3-38) | 36 |
| (41-74)=(41-74) | 34 |
| (134-196)=(125-187) | 63 |
| (202-313)=(193-304) | 112 |
| (316-373)=(307-364) | 58 |
| (378-383)=(369-374) | 6 |
| (385-387)=(376-378) | 3 |
| (396-399)=(387-390) | 4 |
Одинаково выровненныe колонки, не входящиe в блоки отсутсвуют
По результатам оценки программ множественного выравнивания на BAliBASE референсной программой был выбран MAFFT, так как эта программа имеет приемущество над другими выбранными программами по достоверности и скорости обработки данных.
При сравнении с программой MUSCLE получилось самое большое число совпадающих блоков - 9, с программой ClustalO минимальное - 5, с T-Coffee - 8.
По длине выравнивания у программы MUSCLE значения ближе всего к программе MAFFT.(MAFFT-404, MUSCLE - 398,ClustalO - 393, T-Coffee - 395)
Вероятнее всего программы MUSCLE и MAFFT более близкие исходя из полученных данных с помощью программы. Возможно причина этому схожие алгоритмы, лежащие в основе этих программ(программы MAFFT и MUSCLE основаны на итеративном алгоритме)
Задание 3
Для анализа я выбрала семейство ДНК-связывающего домена B3 (B3 DNA binding domain; AC - PF02362), которое было использовано в практикуме 11.
Выбраны были следующие белки из Arabidopsis thaliana - 1YEL, 5OS9, 7ET6.
С помощью TM-align алгоритма на сайте PDB было построено выравнивание по совмещению структур, причем белок 1YEL считался референсным.
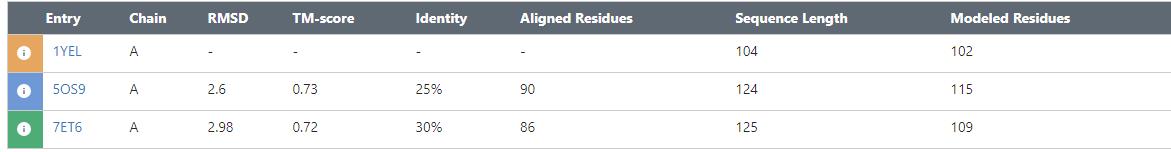 >
>

В проекте Jalview(по ссылке ниже) можно сравнить PDB-выравнивание и множественное выравнивание последовательностей с помощью программы MAFFT.
У PDB выравнивания лучше всего выровнились между собой части с N-конца, с C-конца схожих участков практически нет.Если обратиться к множествнному выравниванию, то 1YEL хуже выровнился относительно других белков(а он как раз и был выбран референсным для двух выравниваний в PDB).
Задание 4. Описание программы MAFFT
MAFFT - программа для создания множественных выравниваний аминокислотных или нуклеотидных последовательностей.Согласно результатам сравнения программ на BaliBase MAFFT обладает высокими показателем точности, а так же сравнительно небольшим временем работы, что является немаловажным фактором. Сочетание этих свойств делает MAFFT мощным и популярным инструментом для множественного выравнивания. В основе программы MAFFT лежит итеративный алгоритм.
Алгоритм прогрaммы:
1.Попарное выравнивание (на этом этапе для всех пар последовательностей строится парное выравнивание)
2.Построение матрицы расстояний (вес парных выравниваний используется для построения матрицы расстояний - больший вес соответствует меньшему расстоянию между последовтаельностями)
2.Построение направляющего дерева( с использованием матрицы расстояний строится направляее дерево, которое отражает сходство между последовательностями)
3.Прогрессивное выравнивание(выполняется от листьев к корню дерева)
4.Итеративное уточнение(повторяет процесс с корректировкой положения гэпов и вставок для повышения точности выравнивания)
Использованные источники:
2.Лекция