Нуклеотидный blast
Сравнение списков находок нуклеотидной последовательности 3-я разными алгоритмами blast
Есть нуклеотидная последовательность, (созданная при обработке хроматограммы с секвенатора). Следует посмотреть, как различные алгоритмы blast будут искать подходящие (гомологичные) последовательности для исходной, чтобы понять, когда лучше какой использовать. Для этого нужно уменьшить количество находок. Это было получено ограничением поиска blast внутри рода лучшей находки - Polycirrus. Так же ограничение на количество находок было 5000 последовательностей.
При запуске Blast с разными алгоритмами были получены следующие результаты:
- blastn. Всего было выдано 34 находки. 27 из них были последовательности, кодирующие cytochrome oxidase subunit 1 (COI), а 7 - рибосомальную РНК
(несмотря на то, что процент идентичности там 100) программа нашла кусочек последовательности длиной 11 и
выровняла его с исходной. Эти семь "последовательностей" далее
ни в каких списках находок не присутствуют. Поэтому E-value худшей находки равен 2.5, но лучшая по-прежнему с e-value 0.0.
Из сказанного выше следует, что этот алгоритм ищет все возможные выравниваемые последовательности с длиной не меньше 11 нуклеотидов (параметр).
Подойдет для выравнивания последовательности (а значит и для поиска ее неизвестной функции и принадлежности организма ее содержащего к систематической категории),
о которой мы ничего не знаем. Что-нибудь да найдем.

Рис.1 Таблица находок, выданная алгоритмом Blastn. - discontiguous megablast. Всего было найдено 27 последоватлеьностей. Все они были в предыдущей выдаче. Теперь e-value наихудшей находки стал 2e-147.
Наилучшая осталась прежней.
Этот алгоритм отсекает неинформативные выравнивания и оставляет наиболее значимые.
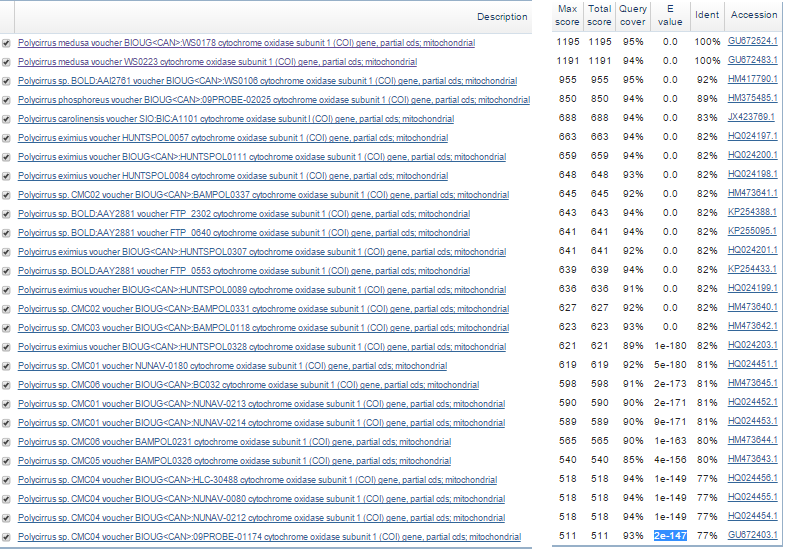
Рис.2 Таблица находок, выданная алгоритмом discontiguous megablast. - megablast Оставил первые четыре находки. E-value "наихудшей" стал 0.0. Отобрал самых близких гомологов среди последовательностей.
На этом этапе можно отследить отдельные нуклеотидные замены у различных видов или представителей одного.
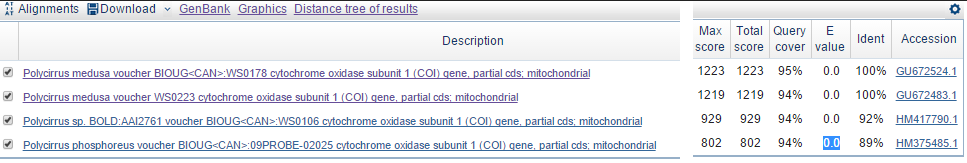
Рис.3 Таблица находок, выданная алгоритмом megablast.
При повтором запуске всех трех алгоритмов, но теперь уже с ограничением по семейству Terebellidae произошла, на мой взгляд интересная выдача: blastn выдал на 2 последовательности меньше (106), чем discontiguous megablast (108). Причем прежних находок с очень плохим e-value (последовательности, кодирующие рибосомальные РНК ) не было. Зато новыми были выданные discontiguous megablast две последовательности опять маленькой длины, но с хорошим процентом сходимости. Они, надо сказать, кодируют тот же белок, что и query. Вывод: на данном примере стало понятно, что discontiguous megablast выдает последовательности (пусть даже с неочень хорошим выравниванием) кодирующие тот же белок (или другие функционально важные элементы), что и исходная последовательность. Этот алгоритм может быть полезен для поиска схожих последовательностей ( а значит и их продуктов) в самых разных организмах. Megablast по-прежнему оставляет наиболее идентичные последовательности.
Поиск гомологов пяти белков в геноме X5 (Amoboaphelidium).
C помощью локального blast была создана нуклеотидная база данных из файла сборки X5 (Amoboaphelidium). С помощью TBLASTN, по базе данных, транслируемой в шести рамках были найдены гомологи следующих белков ( в скобках указаны индентификаторы Uniprot) : Peroxiredoxin(Q9NL98), Dynein heavy chain, cytoplasmic (P37276), Mitochondrial ribonuclease P protein 1 (Q3UFY8), Eukaryotic translation initiation factor 3 subunit G-1 (Q9W4X7), Transmembrane protein 144 homolog A(Q55FV8). Выбор белков основывался на существовании гомологов среди всех эукариот.
- Peroxiredoxin
Пероксид водорода относится к реактивным формам кислорода и при повышенном образовании в клетке вызывает оксидативный стресс.
Это яд для клетки. Правда, некоторые орагнизмы научились его применять как бактерицидное средство.
Выбранный белок является ферментом, устраняющим перекись, вырабатываемую в ходе метаболизма. Это антиоксидант.
В файле,
возвращенном Blast, имеется информация о трех потенциальных гомологах данного белка, найденного в транслированном (в шести рамках) геноме.
Для лучшей находки: SubjectAccession = scaffold-423, Score = 251 bits (642), Expect = 6e-76, Identities = 120/193 (62%), Positives = 151/193 (78%),
Gaps = 2/193 (1%), QueryCover = 98
Так же можно заметить, что первые две находки резко отличаются по значениям параметров от третьей.
Третья находка из-за большого количества гэпов точно не является нужной.
Поэтому возможными гомологами белка пероксиредоксина будут только первая и вторая находки. Почему их две?
При просмотре последовательностей становится понятно, что это две разные последовательности (нет перекрывания между скэффолдами). Они имеют схожие
(возможно консервативные) участки, где абсолютно идентичны. Предположение: ввиду того, что пероксиредоксиныбывают нескольких видов
( в зависимости от количества цистеина в активном центре - собственно тиольная(-sh) группа цистеина и окисляется перекисью.
Возможно в геноме закодировано несколько видов пероксиредоксина. Дополнительные данные в форме таблицы с нужными колонками можно посмотреть
здесь.
- Dynein heavy chain, cytoplasmic
Это цитоплазматический (бывают и аксонемные) белок динеина. Это очень интересные белки! Они называются моторными.
Их функция - превращение энергии АТФ в механическую энергию. Они способны перемещаться по поверхности микротрубочек, перенося грузы (везикулы и органеллы).
В файле выдачи представлена только одна убедительная находка (легко определяется по длине выравнивания).
Несмотря на то, что показатели лучшей находки не очень хорошие (SubjectAccession = unplaced-816, Score = 2157 bits (5589), Expect = 0.0, Identities = 1402/4264 (33%), Positives = 2326/4264 (55%), Gaps = 270/4264 (6%) QueryCover = 90 ,
все равно попадаются консервативные места в большом выравнивании. Это позволяет утверждать, что в данном геноме закодирован гомолог (пусть даже и не очень близкий)
этого моторного белка
(которые присутствуют во всех клетках эукариот). Дополнительные данные в форме таблицы с нужными колонками можно посмотреть
здесь.
- Eukaryotic translation initiation factor 3 subunit G-1
Субъединица G-1 эукариотического третьего фактора инициации трансляции. Является компонентом комплекса, участвующего в трансляции.
Обеспечивает связывание мРНК и methionyl-tRNAi к 40s субъединице рибосомы. Blast выдал следующий
файл. Здесь четко проглядываются 2 находки. Но при просмотре последовательностей,
становится очевидно, что это одна и та же последоватльность, содержащаяся в двух перекрывающихся скэффолдах. Значение параметров для этих выравниваний одинаковые:
SubjectAccession = scaffold-20, Score = 98.2 bits (243), Expect = 2e-22,
Identities = 85/287 (30%), Positives = 127/287 (44%), Gaps = 49/287 (17%) QueryCover = 90 . Выравнивание не особо показательно относительно гомологии,
но все-таки я считаю,
что последовательность, полученную трансляцией участка генома можно считать гомологичной исходной.
(После заключительной последовательности из гэпов выравнивание заметно более лучше). Дополнительные данные в форме таблицы с нужными колонками можно посмотреть
здесь.
- Transmembrane protein 144 homolog A Трансмембранный белок. Функция была не указана. (предположительно, транспорт веществ через мембрану) Blast выдал следующий файл. Значения параметров для лучшей находки: SubjectAccession = scaffold-20, Score = 31.6 bits (70), Expect = 0.81, Identities = 18/61 (30%), Positives = 29/61 (48%), Gaps = 2/61 (3%) QueryCover = 17. Здесь видно, что Blast выровнял одну и туже последовательность , но разной длины (т.е. локальное перекрывание скэффолдов меньше длины выровненного фрагмента последовательности). Из файла можно заметить, что последовательности query гораздо больше по длине, чем участок используемый для выравнивания. Я думаю, что, возможно, Blast нашел только одну или несколько спиралей, гомологичных тем, что в исходной последовательности. Вероятно, для трансмембранных довольно тяжело определить гомологов из-за большого количества структурных элементов (тех же самых спиралей).Дополнительные данные в форме таблицы с нужными колонками можно посмотреть здесь.
- Mitochondrial ribonuclease P protein 1 Фермент, участвующий в созревании митохондриальной тРНК. Вместе с другими состовляющими разрезает тРНК с 5' конца. Blast выдал следующий файл. Значения параметров для единственной находки: SubjectAccession = scaffold-85, Score = 66.6 bits (161), Expect = 1e-11, Identities = 58/209 (28%), Positives = 90/209 (43%), Gaps = 44/209 (21%), QueryCover = 43. Один гомолог данного белка закодирован в геноме, сборка которого была предоставлена. Дополнительные данные в форме таблицы с нужными колонками можно посмотреть здесь.
Классификация геномов родственных вирусов по сходству последовательностей
Для работы был выбран вирус Bat polyomavirus 5a DNA isolate: 5a. Он является ДНК-содержащим и относится к роду Polyomavirus . Все представители этого рода (отсюда и название) могут вызывать многочисленные опухоли у птиц и млекопитающих, в том числе и у человека. В данном случае вирус был найден у летучей мыши.
Для анализа сходства были выбраны следующие геномы:
- Bat polyomavirus 5a DNA isolate: 5a - NC_026768.1
- Bat polyomavirus 5b DNA isolate: 5b-1 - NC_026767.1
- Bat polyomavirus 6a DNA isolate: 6a - NC_026762.1
- Bat polyomavirus 6b DNA isolate: 6b - NC_026770.1
- Bat polyomavirus 6c DNA isolate: 6c - NC_026769.1
- Bat polyomavirus 6d DNA isolate: 6d-1 - NC_026766.1
Затем все эти геномы были помещены в один файл. C помощью makeblastdb
была создана база данных индексов для blast.
С помощью ниже представленной команды была получена
таблица с информацией о выравнивании трансляций всех против всех.
tblastx -query all_viral_fasta.fasta -db db_viral.fasta -out viral_blast.out -outfmt 7Чтобы убрать ненужную информацию из таблицы (например, выравнивание последовательности против себя самой или выравнивание последовательностей А и Б, если уже встретилось выравнивание Б и А) был использован следующий скрипт.
python viral_script.py -i viral_blast.out -s 40 -l 30 -e 1 -b 30 -o viral_script_handle.txtРезультат здесь.
Далее нужно было попытаться проанализировать данные таблицы, определить наиболее и наименее сходные геномы и, соответственно, класcифицировать их.
Первое, что я сделала, это посмотрела структуру генома всех вирусов. Она оказалась схожей (см. рис.4). Кольцевая двуцепочечная ДНК. Всего 4 гена. Первые два кодируют белки капсида и лежат на прямой цепи. Далее идет ген большого Т-антигена - белка, который играет важную роль в регуляции жизненного цикла вируса, связываясь с вирусной ДНК, и усиливая ее репликацию, так как активизирует процессы репликации в самой клетке. Он лежит на комплементарной цепи. Там же лежит ген малого Т-антигена, который активирует клеточные пути, стимулирующие пролиферацию клеток. Только есть особенность: внутри гена, кодирующего белок большой Т-антиген есть некодирующая последовательность. Через несколько нуклеотидов после ее начала лежит второй ген, кодирующий малый Т-антиген. Это пример того, как в организме с малым размером генома гены расположены очень компактно. Мною был выбран вирус специально с небольшим размером генома, чтобы потренироваться на методах оценки сходства вирусных геномов(возможность вручную перебрать находки). Вирусы очень быстро эволюционируют. Поэтому для оценки их родства я выбрала гены, которые, по моему мнению, должны быть наиболее схожи и статичны у представителей одного вида. Это ген большого Т-антигена и ген малого Т-антигена.
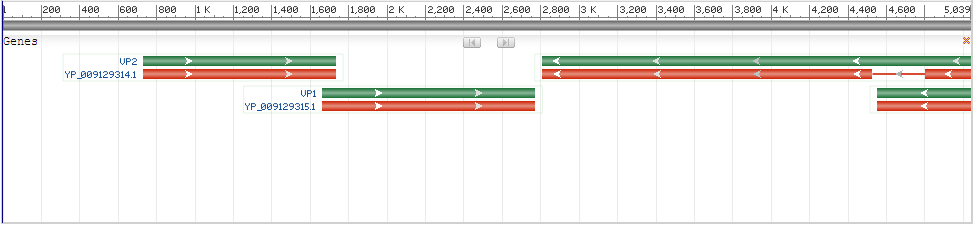
Рис.4 Расположение генов в геноме NC_026766.1
| NC_026766.1 | NC_026767.1 | NC_026768.1 | NC_026769.1 | NC_026762.1 | NC_026770.1 | |
| Координаты гена большого Т-антигена | 4956-2686 | 5047-2637 | 5075-2629 | 5046-2742 | 5019-2775 | 5039-2775 |
| Длина гена большого Т-антигена | 661 | 626 | 647 | 674 | 641 | 652 |
| Координаты гена малого Т-антигена | 4956-4456 | 5047-4309 | 5075-4665 | 5046-4549 | 5013-4531 | 5039-4551 |
| Длина гена малого Т-антигена | 166 | 188 | 136 | 165 | 162 | 162 |
- Таблица была отсортирована по координатам начала query по убыванию. Следовательно, в начале таблицы у меня были находки из сразу двух нужных генов
- Определив порог в координатах нужного гена, я выделила нужное количестов строк (находок) в таблице.
- Далее я отсоритировала таблицу последовательно по полю query и по полю subject. Получилось довольно удобная табличка по парам геномов.
- Далее я определяла наиболее схожие и сравнительно длинные участки. При разборе выявилась закономерность, повторяющаяся у все пар геномов, кроме одной
(NC_026767.1 и NC_026768.1).
- В конце последовательности гена наблюдается сходный участок(либо их несколько) (кодирует 60-80 аминокислот - это меньше, чем последовательность малого Т-антигена, т.е. какая-то его часть). Далее идут много таких же, но хуже выровненных. Я выбирала этот первый участок, так как его координаты конца совпадали с координатами конца гена малого Т-антигена (см. таблицу 1)
- Затем идет длинный консервативный участок
- Затрагивающий начало большого Т-антигена участок (или несколько) также хорошо выровнен у пар геномов.
Все замеченные участки в таблице выделены разными цветами: начальный - розовым, срединный - ярко-зеленым, терминальный - синим. Каждый геном размечен своим цветом.
Отдельно нужно сказать про пару NC_026767.1 и NC_026768.1. НАходки при выравнивании участков из этих двух геном разительно отличаются от тех, что встречались прежде. Их больше, но они более раздроблены (нет длинного среднего участка). Проценты идентичности у этих участков больше, чем у других пар. Очевидно, что эти два генома резко отличаются от остальных. В таблице параметры выравнивания с их участием выделены серым. Точно можно скзать, что эти два генома обособлены от других, однако не теряют связи (есть довольно хорошо выровненные участки и с другими геномами). Они либо быстрее эволюционировали, либо наоборот, изменились в меньшей степени.
Далее я сравниваю первые участки у остальных пар геномов. Понимаю, что их различие не показательны. Поэтому решаю не учитывать их при оценке сходства. Средний участок разительно отличается по показателям (главным образом, по длине). Это и будет основным критерием. Различия по выравниваниям терминальных участков в точности повторяют различия средних участков.
Результаты
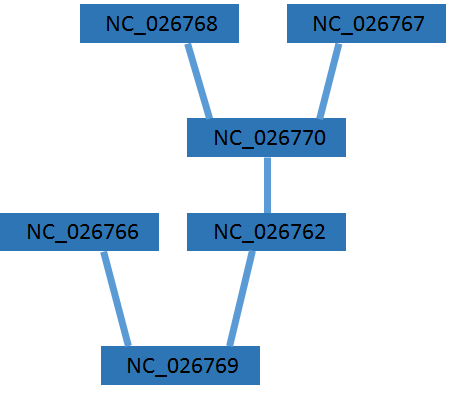
По показателям средние участки были классифицированы следующим образом. (в скобочках указано значение BitScore)
| Сильное сходство | Слабое сходство |
| NC_026762.1 и NC_026770.1 (861) | NC_026762.1 и NC_026767.1 |
| NC_026766.1 и NC_026762.1 (830) | NC_026767.1 и NC_026770.1 |
| NC_026766.1 и NC_026770.1 (761) | NC_026768.1 и NC_026770.1 |
| NC_026766.1 и NC_026769.1 (614) | NC_026766.1 и NC_026767.1 |
| NC_026762.1 и NC_026769.1 (611) | NC_026762.1 и NC_026768.1 |
| NC_0267670.1 и NC_026769.1 (595) |
Исходя из этих данных была составлена следующая схема.
© 2014 Макарова Надежда