|
Третий семестр
Поиск сигналовСравнение состава систем рестрикции модификации, закодированных в двух штаммах вида Ethanoligenens harbinense .Для сравнения был взят полный геном из NCBI (идентификатор: CP002400.1) и набор контигов из метагенома кишечника человека. Последовательности:
Для определения того, какие из имеющихся сайтов присутствуют у интересующей бактерии, производился подсчет контраста: отношение наблюдаемой частоты встречаемости сайта к ожидаемой. Так как известно, что, несмотря на то, что сайты рестрикции в бактериальном геноме метилированы и не должны разрезаться, иногда случаются ошибки, поэтому сайты систем рестрикции-модификации должны встречаться в бактериальном геноме реже ожидаемого. В данном случае подсчет контраста производился по методу Карлина, а пороговое значение контраста было определено в 0.78 (т. е. значение контраста такое и меньше свидетельствует о присутствии данной системы РМ у бактерии). Подсчет контраста выполнялся с помощью веб-вервиса.
После фильтрации по порогу 0.78:
Результат представлен в таблице Excel . Бледно-желтым цветом выделены сайты из полного генома из NCBI с контрастом меньше 0.78 Ярко-желтым выделены сайты, которые встречаются в обоих геномах. Бледно-голубым цветом выделены сайты из набора контигов из метагенома с контарстом меньше 0.78. Ярко-синим выделены сайты, которые встречаются в обоих геномах. Что можно сказать про данные результаты:
Поиск последовательностей Шайн – Дальгарно (SD) в геноме бактерии Amycolatopsis orientalis HCCB10007 .Обзор литературыВикипедия: Последовательность Шайна — Дальгарно (англ. Shine-Dalgarno sequence, Shine-Dalgarno box) — сайт связывания рибосом на молекуле мРНК прокариот, обычно на расстоянии около 10 нуклеотидов до стартового кодона AUG.[1] Описана австралийскими учёными Джоном Шайном и Линн Дальгарно.[2] Консенсусом является последовательность из шести нуклеотидов AGGAGG; в случае E. coli последовательность Шайна — Дальгарно — AGGAGGU. Комплементарная последовательность CCUCCU, называемая последовательностью анти-Шайна — Дальгарно, располагается на 3'-конце молекулы 16S рибосомной РНК. Комплементарное взаимодействие между последовательностями Шайна — Дальгарно и анти-Шайна — Дальгарно служит для помещения старт-кодона мРНК в P-сайт рибосомы для начала биосинтеза белка.[3] При поиске в PubMed по запросу Amycolatopsis orientalis HCCB10007 Shine–Dalgarno была выдана статья [4], где сравнивали 16s рРНК Nocardia lactamdurans делая вывод о ее принадлежности к роду Amycolatopsis , хотя ее раньше относили к Streptomyces. По филогенетическому дереву она довольно близка к нашему виду. В статье приведена последовательности анти-SD:CUCCUU (комплементарная - AAGGAG). Далее статье [5] было указано, на координаты относительно старта трансляции для Streptomyces (-11, -10) Поиск мотива Была взята последовательность хромосомы , а также
features-файл. Далее с помощью скрипта1
были получены координаты кодирующих последовательностей, коих насчиталось 8114.
После отбора только аннотированных генов было выбрано 990 последовательностей с длиной от 1320 до 17151.
Затем были определены границы по координатам областей, где могут находится последовательности Шайна-Дальгарно (с учетом, что трансляция генов на обратной цепи начинается с максимальной координаты).
Далее с помощью скрипта2 были последовательности с желаемыми границами.
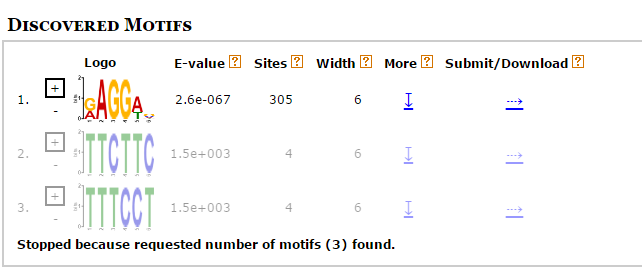
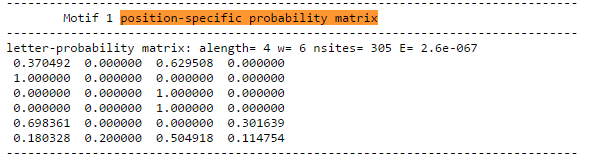
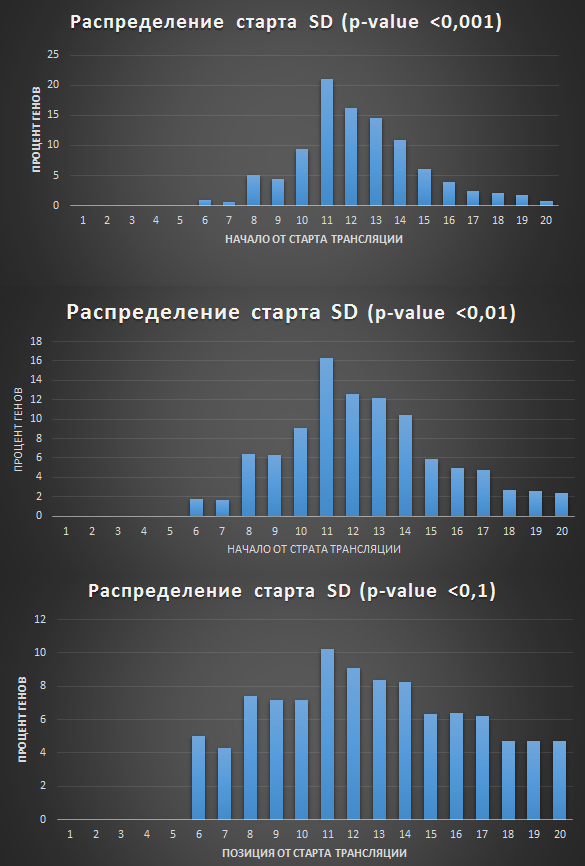
Результаты представлены на Рис.1 Найден мотив, который резко отличается по E-value, а значит более достоверный и является искомой последовательностью Шайна-Дальгарно. Его LOGO представлен на Рис.2. Также была посчитана позиционная матрица весов (PWM), представленная на Рис3. Она необходима, чтобы найти эту последовательность в других генах. С помощью программы FIMO был произведен поиск найденного мотива для всех остальных генов бактерии Для этого были вырезаны участки от -20 до -1 позиции до начала каждой кодирующей последовательности. Рис.1 Три найденных мотива для области -20, -1 кодирующих последовательностей. Рис.2 LOGO найиболее достоверного мотива. Рис.3 Позиционная весовая матрица (PWM) мотива Шайна-Дальгарно, построенная программой MEME. Поиск мотива во всех генахДля 8114 генов было найдено 24082 мотива (с p-value меньше 0.1) Такое большое число находок скорее всего связано с наличием случайных последовательностей, так как сигнал SD достаточно слабый. Среди найденных 1395 последовательностей (5,79%) имело p<0.001, 5647 последовательностей (23%) - p<0.01, 9597 последовательностей (39.8%) имело p <0.03. Так как SD должна быть в большинстве генов, то для анализа был выбран p-value <0.01. Но для сравнения были также получены результаты по p-value <0.001 и <0.1. Результаты представлены в таблице. Были построены гистограммы распределения начала найденных SD от старта трансляции. Полученные изображения представлены на рисунках 4. Как видно, наибольшая часть SD приходится на -11 нуклеотид от начала трансляции (и конец, соответсвенно, на -7), что согласуется с литературными данными. Рис.4 Гистограммы распределения начала найденных SD от страта трансляции для порого p-value 0.001 (1396/8114 - 17,2% генов), p-value 0.01 (5647/8114 - 69.6%), p-value 0.1. Числа на оси X означают расстояние от начала SD до старта трансляции ( по идее, их стоит рассматривать как отрицательные числа, так как SD расположена до начала кодирующей рамки). Также для найденных мотивов было построено лого для p < 0.01 (Рис.5). Оно совпадает с последоватаельностью из статьи, за исключением первой буквы. Лого последовательности Шайна-Дальгарно, построенное по находкам этого мотива в геноме Amycolatopsis orientalis HCCB10007 (для порога p-value 0.01). Изображение получено с помощью сайта LOGO Источники:
Дата последнего изменения: 22.10.14
© 2014 Макарова Надежда |