Упражнения
Для начала создадим файл в USA-формате (listfile) в директории ls. Используем команду ls>list.
Затем объединяем эти файлы в один с помощью программы seqret: seqret @list sequences.fasta
Используем команду seqretsplit completesequences.fasta seq.fasta В файле completesequencea.fasta записано три последовательности. Программа seqretsplit записывает каждую из этих последовательностей в отдельный файл.
На вход подана некая нуклеотидная последовательность мРНК и выбрана таблица генетического кода №5.Программа транслирует последовательность начиная с первого нуклеотида (а не ищет старт-кодон), поэтому имеем множество стоп-кодонов(*).
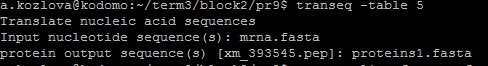


Я использовала файл NC_013943.gbk - запись для хромосомы из бактерии Denitrovibrio acetiphilus DSM 12809

С помощью команды shuffle -o shuffled.fasta sequence.fasta я перемешала буквы в последовательности sequence.fasta и записала полученный результат в файл shuffled.fasta


Почти для всех динуклеотидов частота встречаемости не очень значительно отличается от ожидаемой
Сравнение аннотации генов белков в одной хромосоме бактерии или археи с трансляциями длинных открытых рамок считывания
Для работы я выбрала бактерию из 1 семестра Denitrovibrio acetiphilus DSM 12809, которая имеет одну хромосому.AC (в GenBank CP001968, в RefSeq NC_013943) Последовательность хромосомы в формате genbank
Рис1. Бактерии под увеличением
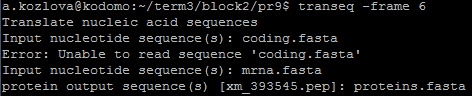
Получение трансляции открытых рамок с помощью команды getorf пакета EMBOSS
Так как данная программа извлекает открытые рамки считывания только с нуклеотидных последовательностей (которых нет в формате .gb), был использована Последовательность хромосомы в формате fasta
Я установила следующие опции
| Опция | Значение |
| -table 11 | Таблица генетического кода для генома бактерии |
| -minsize 180 | Минимальная длина открытой рамки - 180 п.н. |
| -circular | Кольцевая хромосома |
| -find 0 | Выходные последовательности - трансляции открытых рамок от стоп кодона до стоп кодона |
Fasta-файл с открытыми рамками
Получение списка координат и ориентаций найденных открытых рамок с помощью infoseq
| Опция | Значение |
| -only | Показывать только указанные параметры |
| -name | ID открытой рамки |
| -length | Длина трансляции в остатках |
| -description | Описание содержит координаты открытых рамок |
Обработанный результат в формате .xlsx
Для получения списка аннотированных генов белков я скачала файлы с расширениями .ptt (хромосомная таблица со списком генов белков)и .faa (с последовательностями белков в формате fasta):NC_013943.ptt,NC_013943.faa
Обработав файл NC_013943.ptt я получила таблицу аннотированных генов белков в формате Excel:Prots.xslx
Для сравнения двух полученных таблиц я использовала Excel.
Полученная таблица: compare.xlsx
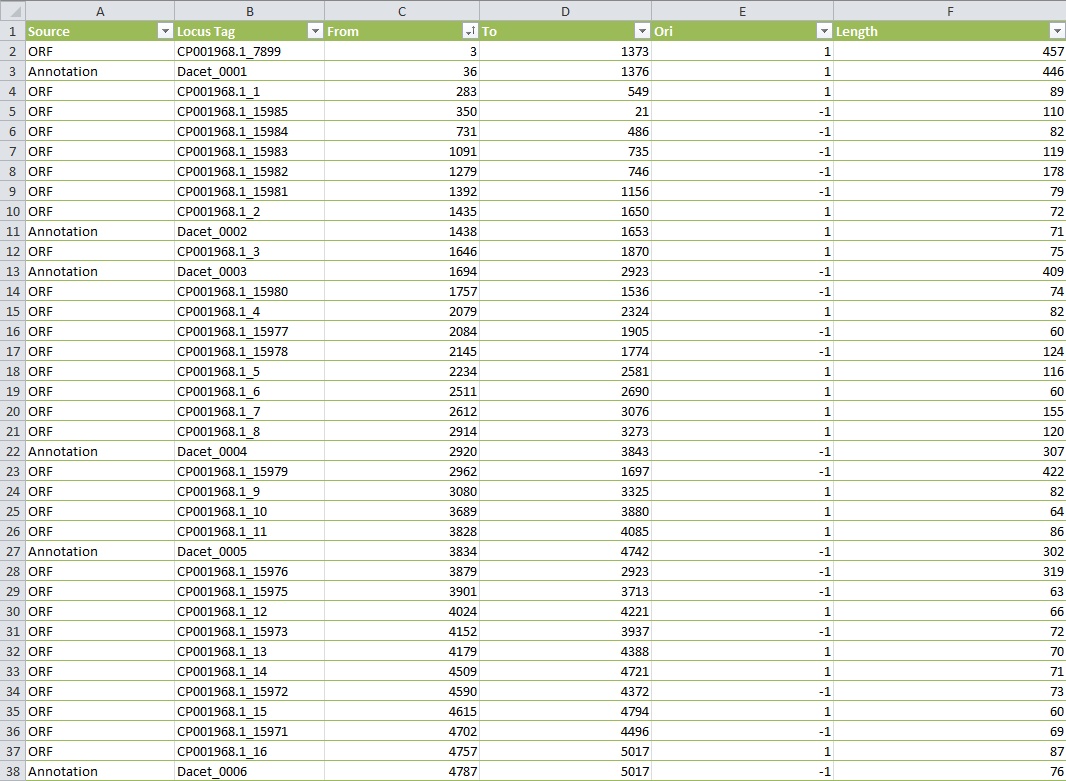
Рис2. Фрагмент сравнительной таблицы
Можно заметить, что открытые рамки длиннее, чем соответствующие белки
Аннотированных белков примерно в несколько раз меньше, чем открытых рамок. Это можно объяснить тем, что наличие открытой рамки не гарантирует присутствие гена, кодирующего белок.
© Козлова Анастасия, 2015