
Файл с протоколом выполнения практикума → novikova_pr5.doc.
Все файлы, полученные при выполнении практикума → ../term2/block2/credits/Pr5
Задание №1
[В этом задании необходимо было получить несколько коротких фрагментов (по 20 аминокислот) из искусственно смоделированного мутанта своего белка (в моем случае – белка THIE_BACSU) при помощи специального скрипта evolve_protein.pl.]
1.
Запустим скрипт evolve_protein.pl из Putty, меняя при этом параметры, отвечающие
за «вероятность изменения остатка» (-c)
и «вероятность замены остатка» (-r):
·
perl
evolve_protein.pl -i P39594.fasta -c 0.6 -r 0.6 -o
seq1.txt
·
perl
evolve_protein.pl -i P39594.fasta -c 0.6 -r 0.8 -o
seq2.txt
·
perl
evolve_protein.pl -i P39594.fasta -c 0.4 -r 0.8 -o
seq3.txt
2.
Получив 3 файла, содержащих мутированные последовательности белка из 20
аминокислот создадим файлы формата .fasta (seq1.fasta,
seq2.fasta,
seq3.fasta),
содержащие fasta исходного белка и мутированной последовательности.
3.
Теперь загрузим получившиеся
fasta-файлы
в
JalView и
настроим цветовую схему для аминокислот следующим образом (Colour>User
defined)):
o
Положительно заряженные –
span lang="EN-US" style="mso-ansi-language: EN-US">Lys,
Arg,
His
– светло-зелёным цветом
o
Отрицательно заряженные –
Asp, Glu – синим цветом
o
Полярные – Ser, Thr, Cys,
Asn, Gln – красным цветом
o
Неполярные
– Gly, Ala, Val, Leu, Ile, Pro, Met – жёлтым
o
Ароматические – Phe, Tyr,
Trp – темно-зелёным
4. Цветовую схему (novikova_colour.jc) сохранили и применили для всех трёх файлов в проекте JalView.
5.
Далее выровняли вручную все три пары последовательностей и в таблице привели описание (%
идентичности, % сходства, вес по матрице
Blosum62
(штраф за открытие пробела (gap) в -12, а за продолжение пробела -2) – считали с
использованием формул ИНДЕКС, ПОИСКПОЗ и СУММ в Excel) для каждого выравнивания:
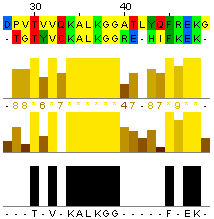
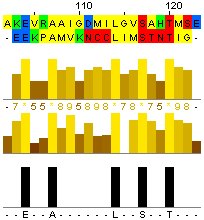
| Выравнивание с мутантом №1 (-c 0.6 –r 0.6) | |||
|
|
% идентичности: (10/21)*100 = 47,6% | ||
| % сходства: (14/21)*100 = 66,7% | |||
| Вес: 34 | |||
| Выравнивание с мутантом №2 (-c 0.6 –r 0.8) | |||
|
|
% идентичности: (5/20)*100 = 25% | ||
| % сходства: (10/20)*100 = 50% | |||
| Вес: 18 | |||
| Выравнивание с мутантом №3 (-c 0.4 –r 0.8) | |||
|
|
% идентичности: (18/20)*100 = 90% | ||
| % сходства: (18/20)*100 = 90% | |||
| Вес: 85 | |||
6. Описание результатов:
· Несмотря на большое различие
между процентами идентичности и сходства в первых двух выравниваниях и,
напротив, совпадение этих значений у третьего выравнивания, объективным
параметром сравнения всё же стоит считать % идентичности. % сходства является
наименее объективным параметром сравнения выравнивания из трёх предложенных
из-за того, что он зависит от выбора групп аминокислот, считающихся схожими в
конкретном случае. Но стоит отметить, что использование % сходства довольно
удобно при сравнении функциональных свойств двух белков.
· Ни по одному из параметров
нельзя сказать, что выравнивания сходны между собой. В последнем выравнивании
из-за того, что вероятность внесения изменения остатка составляла всего 0.4, 90%
аминокислот остались консенсусными для двух последовательностей, что даёт
последнему выравниванию наибольшие результаты по всем трём критериям оценки
качества выравнивания.
Задание №2
[В этом задании необходимо было построить выравнивание своего белка и его предполагаемых ортологов или гомологов.]
1. В качестве ортологов белка
THIE_BACSU были выбраны белки
THIE_BACCQ и
THIE_BACHD (из результатов
предыдущих практикумов).
2. Сохранили их последовательности в общем файле в fasta-формате, объединив информацию из трёх фалов с помощью команды cat:
cat thie_bacsu.fasta thie_baccq.fasta thie_bachd.fasta > thie.fasta
3. Открыли файл с последовательностями в
JalView
и выровняли с помощью программы
Muscle.
4. Сохранили информацию о трёх парах последовательностей
(Selection>Output to text box>Fasta) в
fasta-формате (thie_bacsu_baccq.fasta,
thie_bacsu_bachd.fasta,
thie_baccq_bachd.fasta).
5. С помощью команды infoalign получили данные о попарных выравниваниях:
· infoalign thie_bacsu_baccq.fasta
· infoalign thie_bacsu_bachd.fasta
· infoalign thie_baccq_bachd.fasta
6. Полученные данные перенесли в Excel:
| Name | SeqLen | AlignLen | Gaps | GapLen | Ident | Similar | Differ | % Change | Weight | Description |
| THIE_BACSU/1-222 | 222 | 226 | 3 | 4 | 222 | 0 | 0 | 1.769912 | 1.000000 | Thiamine-phosphate synthase OS=Bacillus subtilis (strain 168) GN=thiE PE=1 SV=1 |
| THIE_BACCQ/1-219 | 219 | 223 | 3 | 4 | 131 | 34 | 54 | 41.255604 | 1.000000 | Thiamine-phosphate synthase OS=Bacillus cereus (strain Q1) GN=thiE PE=3 SV=1 |
| Name | SeqLen | AlignLen | Gaps | GapLen | Ident | Similar | Differ | % Change | Weight | Description |
| THIE_BACSU/1-222 | 222 | 226 | 3 | 4 | 222 | 0 | 0 | 1.769912 | 1.000000 | Thiamine-phosphate synthase OS=Bacillus subtilis (strain 168) GN=thiE PE=1 SV=1 |
| THIE_BACHD/1-211 | 211 | 217 | 2 | 6 | 79 | 43 | 89 | 63.594471 | 1.000000 | Thiamine-phosphate synthase OS=Bacillus halodurans (strain ATCC BAA-125 / DSM 18197 / FERM 7344 / JCM 9153 / C-125) GN=thiE PE=3 SV=1 |
| Name | SeqLen | AlignLen | Gaps | GapLen | Ident | Similar | Differ | % Change | Weight | Description |
| THIE_BACCQ/1-219 | 219 | 223 | 3 | 4 | 219 | 0 | 0 | 1.793722 | 1.000000 | Thiamine-phosphate synthase OS=Bacillus cereus (strain Q1) GN=thiE PE=3 SV=1 |
| THIE_BACHD/1-211 | 211 | 217 | 2 | 6 | 92 | 38 | 81 | 57.603687 | 1.000000 | Thiamine-phosphate synthase OS=Bacillus halodurans (strain ATCC BAA-125 / DSM 18197 / FERM 7344 / JCM 9153 / C-125) GN=thiE PE=3 SV=1 |
6. Раскрасили выравнивание по цветовой схеме ClustalX и выбрали порог identity treshold (опция Above identity Threshold) равным 50, чтобы окрашивались только позиции, в которых, как минимум, две совпадающие буквы:
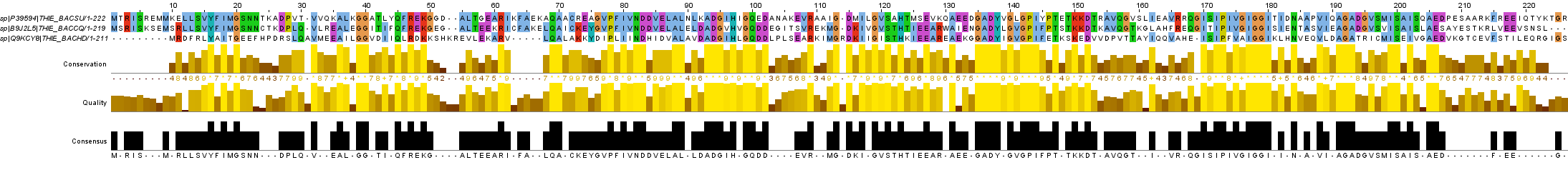
7. Сохранили
проект JalView
с двумя открытыми окнами.