Сборка de novo
В этом задании было необходимо поработать с данными проекта по секвенированию бактерии Buchnera aphidicola.
 |
| Buchnera aphidicola в клетке хозяина, [1] |
Работать было нужно с короткими (длины 36) чтениями, полученными по технологии Illumina. Мне достался код доступа SRR4240357.
На странице проекта я скачала fastq - файл в виде архива .gz и перенесла его
в рабочую директорию (/nfs/srv/databases/ngs/ann_karpukhina1/pr15), где распаковала
программой gunzip.
Использованная команда: gunzip SRR4240357.fastq.gz
Был получен файл с чтениями SRR4240357.fastq.
Очистка чтений
С помощью программы Trimmomatic
была проведена очистка чтений, а именно: удаление остатков адаптеров и плохих букв с концов.
Для начала все адаптеры для Illumina были собраны в единый файл
adapters.fasta.
Затем были исполнены следующие команды:
| Команда | Назначение | Выходной файл |
|---|---|---|
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240357.fastq SRR4240357_noad.fastq ILLUMINACLIP:adapters.fasta:2:7:7 | Удаление остатков адаптеров | SRR4240357_noad.fastq |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240357_noad.fastq SRR4240357_trim.fastq TRAILING:20 MINLEN:30 | Обрезка с концов чтений нуклеотидов с качеством ниже 20 и отбор чтений длины не менее 30 | SRR4240357_trim.fastq |
Выдача программы на этапе удаления адаптеров: Input Reads: 8098979 Surviving: 7937705 (98,01%) Dropped: 161274 (1,99%).
Было отброшено 161274 чтения из 8098979 исходных, размер файла уменьшился с 863 Мбайт до 845 Мбайт.
Выдача программы на этапе обрезки концов и отбора по длине: Input Reads: 7937705 Surviving: 7248100 (91,31%) Dropped: 689605 (8,69%).
Было отброшено 689605 чтений из 7937705, размер файла уменьшился с 845 Мбайт до 748 Мбайт.
Подготовка k-меров
Подготовка k-меров была произведена с помощью программы velveth. Она предназначена для
создания набора данных, которые далее могут обрабатываться программой velvetg.
Velveth принимает на вход несколько последовательностей, строит хэш-таблицу и создает в
отдельной директории два файла - Sequences и Roadmaps, необходимые для velvetg.
При запуске необходимо указывать длину k-мера (hash length) - длину хэшируемых слов в парах
оснований, а также тип чтений (короткие или длинные, парные или непарные и т.д.). Возможные
форматы входных файлов - fasta (default), fastq, fasta.gz, fastq.gz, sam, bam, eland, gerald.
В нашем случае было необходимо подготовить k-меры длины 29 для коротких непарных чтений (-short) из
файла в формате fastq (-fastq). Выходные файлы записывались в папку velveth.
Использованная команда: velveth velveth 29 -fastq -short SRR4240357_trim.fastq.
Cборка на основе k-меров
Cборка на основе k-меров была произведена программой velvetg с использованием данных, полученных
на предыдущем этапе. Velvetg строит граф де Брёйна - ориентированный n-мерный граф из m символов,
отражающий пересечения между последовательностями символов. Он имеет m^n вершин, состоящих из
всех возможных последовательностей длины n из данных символов. Один и тот же символ может встречаться
в последовательности несколько раз.
Запуск программы без дополнительных параметров позволят получить fasta-файл с контигами и
статистические данные в указанной директории.
Использованная команда: velvetg velveth.
В постороенном программой графе оказалось 899 вершин. Информация по каждой из них
отражена в файле stats.txt. Стоит отметить, что количество вершин не обязательно соответствует
количеству контигов, так как "нормальными" являются только контиги длины не менее 29.
Именно они прописываются в файле contigs.fa. В нашем случае их 174. Статистические данные об этих
контигах представлены в файле Excel.
| Длины и покрытия самых больших контигов | |||
|---|---|---|---|
| ID | Длина | Покрытие | Последовательность |
| 19 | 56526 | 40,680324 | contig19.fasta |
| 2 | 45901 | 38,391800 | contig2.fasta |
| 13 | 41937 | 36,650929 | contig13.fasta |
N50 = 25027, максимальная длина контига - 56526. Два следующих по размеру контига имеют длины 45901 и 41937.
Типичные значения покрытий выделить довольно сложно, так как они достаточно сильно разнятся. Чтобы найти отклоняющиеся показатели, были вычислены наиболее употребимые средние значения, а также найдены максимальные и минимальные покрытия.
- Среднее арифметическое - 82,47
- Медиана - 15,726453
- Мода - 1,793103
- Максимальные значения покрытий - 522,045455; 515,852941; 507,253012
- Минимальные значения покрытий - 1,551724; 1,655172; 1,793103
| Контиги с аномально большим покрытием | |||
|---|---|---|---|
| ID | Длина | Покрытие | Последовательность |
| 91 | 88 | 522,045455 | contig91.fasta |
| 72 | 34 | 515,852941 | contig72.fasta |
| 58 | 83 | 507,253012 | contig58.fasta |
| 92 | 45 | 505,688889 | contig92.fasta |
В случае контигов с минимальным покрытием я не уверена, можно ли их назвать "аномальными". Их покрытия действительно значительно меньше тех показателей, которые хотелось бы назвать типичными, однако в выборке их слишком много (26 контигов с покрытием < 3).
Анализ
С помощью алгоритма megablastn было проведено сравнение каждого из трех самых длинных контигов
с хромосомой Buchnera aphidicola
(CP009253).
Результаты работы megablastn можно увидеть в таблице ниже.
| Сравнение самых длинных контигов с хромосомой Buchnera aphidicola | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Координаты в геноме | Max score | Total score | Query cover | E-value | Ident | Alignment length | Gaps | Mismatch |
| 19 | 2004-11103 | 5760 | 26048 | 6% | 0.0 | 7225 (78%) | 9217 | 244 (2%) | 1748 (20%) |
| 2 | 153752-161738 | 4741 | 16278 | 4% | 0.0 | 6348 (78%) | 8171 | 270 (3%) | 1553 (19%) |
| 13 | 202390-207661 | 3349 | 17169 | 4% | 0.0 | 4182 (78%) | 5331 | 141 (2%) | 1008 (20%) |
Построенные выравнивания можно назвать достаточно неплохими.
Далее требовалось выполнить аналогичный анализ для двух контигов с аномально большим покрытием.
При запуске megablastn с дефолтными параметрами ни для одного из 4-х описанных ранее "аномальных" контигов выравнивание построено не было, выдавалось сообщение "No significant similarity found". В FAQ программы указано, что одной из возможных причин такой ошибки могут являться слишком короткие query-sequences, что в нашем случае действительно так. В таких случаях разработчики советуют повысить Expect threshold, что и было сделано, однако ситуацию не изменило.
Другой возможной причиной была указана фильтрация участков малой сложности. Я попыталась выполнить поиск без данной фильтрации, но это вновь ничего не изменило.
Таким образом, ни для одного из контигов с аномально большим покрытием построить выравнивание не удалось.
Изменение длины k-мера
В этом задании было необходимо запустить velveth и velvetg, установив
длину слова 25 вместо 29.
Использованные команды:
- velveth velveth2 25 -fastq -short SRR4240357_trim.fastq
- velvetg velveth2
| Длины и покрытия самых больших контигов | |||
|---|---|---|---|
| ID | Длина | Покрытие | Поcледовательность |
| 3 | 10436 | 55.743484 | contig3.fasta |
| 163 | 7958 | 56.516084 | contig163.fasta |
| 70 | 6826 | 55.514503 | contig70.fasta |
По сравнению с предыдущими результатами как N50, так и максимальные длины контигов значительно уменьшились. Это вполне объяснимо, так как при уменьшении хэш-длины должны соответственно уменьшаться и контиги, Покрытие, напротив, растет, хоть и не слишком сильно.
Для данных контигов был запущен megablastn. Результаты приведены в таблице.
| Сравнение самых длинных контигов с хромосомой Buchnera aphidicola | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Координаты в геноме | Max score | Total score | Query cover | E-value | Ident | Alignment length | Gaps | Mismatch |
| 3 | 333222-339010 | 2878 | 3300 | 1% | 0.0 | 4482 (76%) | 5897 | 187 (3%) | 1228 (21%) |
| 163 | 215746-218384 | 1792 | 2136 | 0% | 0.0 | 2118 (79%) | 2676 | 64 (2%) | 494 (19%) |
| 70 | 209294-212231 | 1541 | 2020 | 0% | 0.0 | 2291 (77%) | 2994 | 102 (3%) | 601 (20%) |
По сравнению с предыдущим анализом длины, вес и Query cover выравниваний очевидным образом понизились. Тем не менее проценты совпадений, однонуклеотидных различий и гэпов остались почти такими же. А вот координаты в геноме ни для одного из контигов не совпали и даже не перекрываются.
Сборка SPAdes
Программа SPAdes – St. Petersburg genome assembler - предназначена как для стандартных изолятов, так и
для бактериальных сборок, полученных в результате single-cell MDA sequencing (секвенирование одиночных клеток с использованием метода
амплификации со множественным замещением цепи - Multiple Displacement Amplification). Текущая версия
SPAdes работает с чтениями Illumina или IonTorrent и способна создавать гибридные сборки, используя
чтения PacBio и Sanger. Также программе можно предоставлять дополнительные контиги для использования в качестве
длинных ридов.
Изначально SPAdes была разработана для небольших геномов и тестировалась на наборах данных
по бактериям и грибам, поэтому она не подходит для сборки крупных геномов (e.g. геномов млекопитающих) и метагеномных
проектов.
Существует несколько модулей SPAdes:
- BayesHammer - производит предварительную коррекцию ошибок чтений Illumina, хорошо работает как для стандартных данных, так и для данных single-cell MDA секвенирования.
- IonHammer - то же, что и предыдущий, но для данных IonTorrent.
- SPAdes - итеративный (повторяющийся) модуль для сборки коротких чтений; значения K выбираются автоматически на основании длины ридов и типа данных.
- MismatchCorrector - инструмент, улучшающий показатели по однонуклеотидным несовпадениям и коротким инделям в получающихся контигах и скэффолдах; по умолчанию он выключен, но рекомендуется его использовать.
- dipSPAdes - модуль для сборки диплоидных геномов с высоким уровнем полиморфизмов.
На вход SPAdes принимает файлы формата FASTA и FASTQ, для данных IonTorrent принимается формат BAM. Также возможна обработка архива gzip.
Для начала я попыталась запустить SPAdes в рекомендованном режиме с коррекцией и параметрами по умолчанию c помощью команды spades.py -s SRR4240357.fastq -o spades , но этого сделать не удалось. Программа прервалась, выдав ошибку. Насколько я понимаю, это связано с ограничением памяти, так как SPAdes рабоает с достаточно большими объемами данных.
Далее я попыталась запустить SPAdes с уже очищенным с помощью Trimmomatic файлом и опцией --only-assembler, позволяющей проводить сборку без предварительной коррекции.
Использованная команда: spades.py -s SRR4240357_trim.fastq --only-assembler -o spades
Параметр -s указывает, что во входном файле короткие и непарные риды, -o обозначает выходную директорию.
В первую очередь нужно отметить, что программа работает очень долго (более 3 часов). По умолчанию SPAdes проводит сборки при различных значениях k, начиная с 21. В нашем случае сборки были сделаны только с k=21 и k=33, затем программа закончила работу с сообщением о превышении параметром k длины рида (Value of K (55) exceeded estimated read length (39)), что, собственно, было ожидаемо.
Отчет о работе программы:
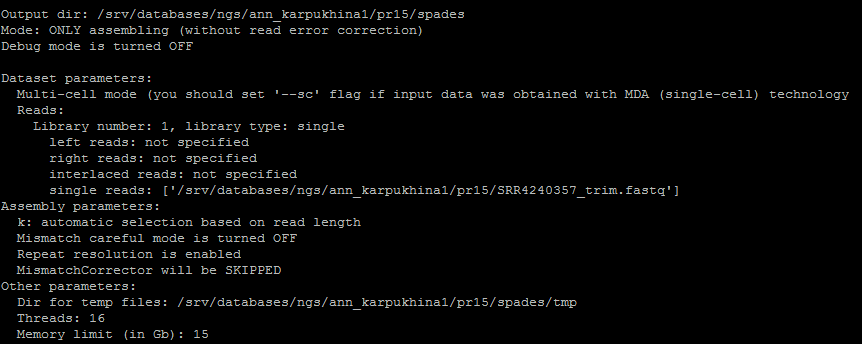
На выходе было получено множество файлов, некоторые из которых были собраны в отдельные папки. Из всего этого нас больше всего интересуют файлы contigs.fasta и contigs.fastg, содержащие последовательности контигов и информацию о них. Аналогичные файлы есть и для скэффолдов (scaffolds.fasta, scaffolds.fastg).
Всего SPAdes построил 75 контигов, что значительно меньше, чем velvetg (174 при k=29).
Обработанные данные по контигам можно найти в файле spades_contigs.xlsx .
| Длины и покрытия самых больших контигов | ||
|---|---|---|
| ID | Длина | Покрытие |
| 1 | 163720 | 16,9567 |
| 3 | 113104 | 17,6205 |
| 5 | 101515 | 19,3695 |
Тем не менее основной параметр, по которому в данном случае можно сравнить качество сборки - N50 - повышается, причем значительно. Для SPAdes N50 = 101515, что более, чем в 4 раза выше соответствующего значения для velvetg. Кроме того, SPAdes проводил сборки сразу для нескольких значений k. Поэтому, на мой взгляд, эта программа работает точнее.