Построение профиля подсемейства
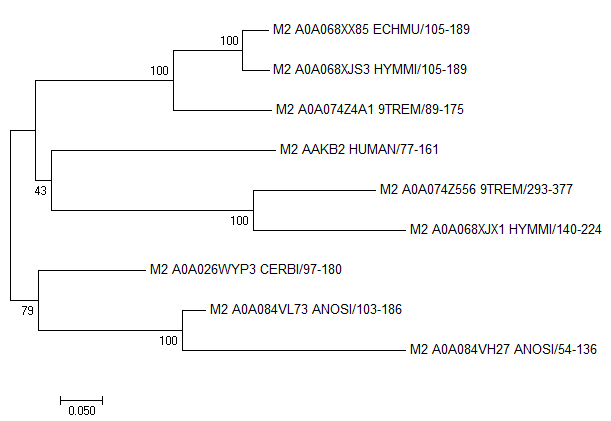 |
| Рисунок 1. Дерево представителей "хорошего" подсемейства, построенное методом Neighbor-Joining c Bootstrap-тестом (100 реплик) |
Для построения и калибровки профиля применялся пакет HMMER, установленный на kodomo.
Использовались следущие команды:
Построение профиля: hmm2build profile1 profile.fasta
Калибровка:hmm2calibrate profile1
Полученный файл: profile1
После этого был произведен поиск по всем белкам UniProt, содержащим домен AMPK1_CBM.
Использованная команда: hmm2search profile1 PF16561_full_length_sequences.fasta >> output
Полученный файл: output
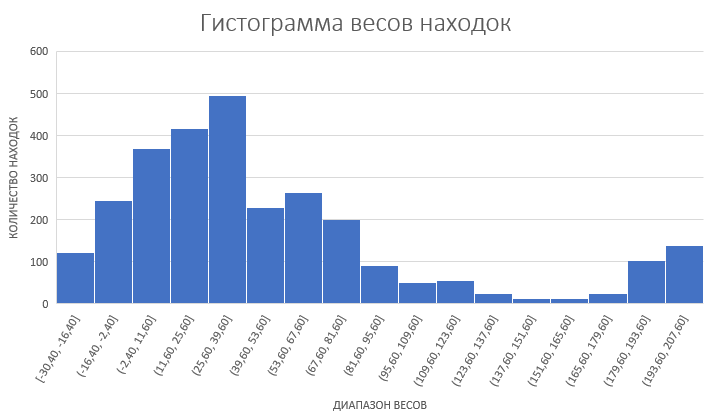 |
| Рисунок 2. Гистограмма весов находок |
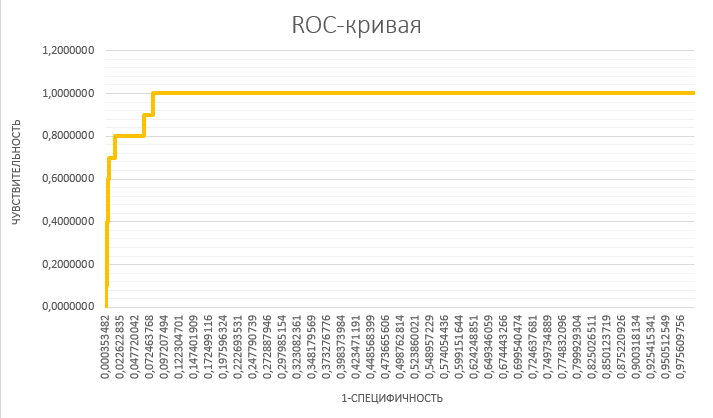 |
| Рисунок 3. ROC-кривая |
По имеющимся данным были построены гистограмма весов находок (Рис. 2) и ROC-кривая (Рис. 3).
Итоговый Excel-файл: roc.xlsx
На основании построенной ROC-кривой было выбрано пороговое значение E-value. Критерием выбора являлось максимальное значение разности [Чувствительность - (1-Специфичность)]. Полученный порог E-value - 1,3E-51. При таком пороге чувствительноть профиля составляет 1,0000000, а специфичность 0,92117356. Это достаточно хорошие значения, поэтому построенный профиль вполне можно использовать для выделения подсемейства.
Далее все имеющиеся находки были разделены по принципу выше/ниже порога, и среди каждой группы были выделены представители, принадлежащие и не принадлежащие подсемейству. Полученные результаты представлены в таблице 1.
| На самом деле | Принадлежит подсемейству | Не принадлежит | Сумма |
| Выше порога по профилю (включая порог) | 9 | 224 | 233 |
| Ниже порога | 0 | 2606 | 2606 |
| Сумма | 9 | 2830 | 2839 |
| Таблица 1. Разделение находок при пороге E-value 1,3E-51 | |||
Поиск гомологов с помощью psi-BLAST
PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) предназначен для
сравнения изучаемой аминокислотной последовательности белка с имеющейся базой данных с целью
поиска последовательностей, обладающих незначительным сходством. Его алгоритм состоит
из множества итераций, в каждой из которых из списка последовательностей
строится множественное выравнивание, из котрого затем извлекаются блоки для построения PSSM
(Position-Specific Scoring Matrix).
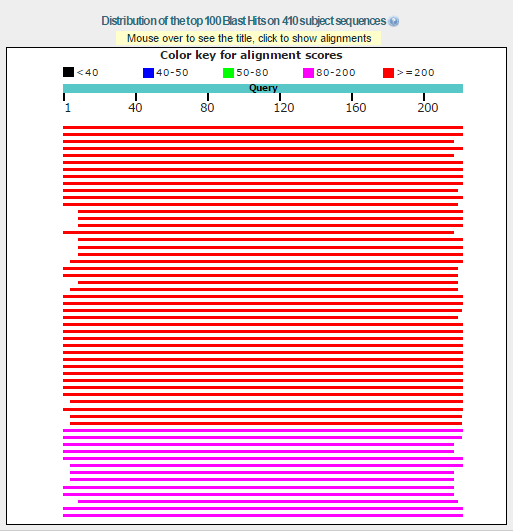 |
| Рисунок 4. Число находок PSI-BLAST после первой итерации |
 |
| Рисунок 5. Выдача PSI-BLAST после последней итерации |
Для данного белка был запущен PSI-BLAST с дефолтными параметрами по банку SwissProt. Сервис сработал достаточно быстро и после первой итерации выдал 410 находок (Рис. 4), из которых 111 имели E-value лучше порогового, а 299 - хуже. E-value худшей из "хороших" находок составил 0.004, а лучшей из "плохих" - 0.005, что согласуется с заданными по дефолту параметрами (PSI-BLAST Threshold = 0.005). E-value самой хорошей находки - 2e-162. Все 111 "хороших" последоваетльностей были выбраны для PSI-BLAST (галочка в поле Select for PSI blast), но ни одна из находок не была использована для построения PSSM (нет галочки в поле Used to build PSSM), что логично, так как это была только первая итерация.
Затем с выбранными "хорошими" находками была запущена вторая итерация, опять же с дефолтными параметрами (Run PSI-Blast iteration 3 with max 500). При этом, как и было задано, нашлось 500 последовательностей, все из них были выбраны для PSI-BLAST, но далеко не все из них были отмечены зеленой галочкой как "правильные" и использованы для построения PSSM. Желтым цветом были отмечены последовательности с весом ниже порогового на основании предыдущей итерации. Последовательностей, использованных для построения PSSM оказалось 109, лучшая из них имела E-value 8e-120, а худшая - 2e-20.
После третьей итерации из 500 находок уже только 12 были выделены желтым, а 488 - использованы для построения PSSM. Худший E-value использованной находки - 7e-54, лучший - 9e-100.
После четвертой итерации желтым оказались выделены лишь 7 находок из 500, а для PSSM использовались 493. E-value худшей из них - 4e-62, лучшей - 2e-101.
После пятой итерации желтым были выделены 2 находки. Для PSSM исползовались 488, среди них лучший E-value - 4e-109, худший - 6e-61.
Шестая итерация вопреки моим ожиданиям выдала больше "плохих" находок - 6. Соответсвенно для PSSM были испоьзованы 494. Худший E-value из использованных находок - 6e-61, то есть такой же, как и в предыдущей итерации. Лучший - 6e-108.
После седьмой итерации число выделенных желтым находок опять увеличилось до 12, для PSSM были использованы 488 находок, худший E-value из них - 6e-66, лучший - 1e-107.
Восьмая итерация - вновь увеличение числа "плохих" находок - 18. Для PSSM использованы 482, из них худший E-value - 6e-70, лучший - 4e-109.
На данном этапе можно заметить следующую тенденцию: E-value худшей из использованных для PSSM находки для каждой последующей итерации становится ниже. Что-то однозначно сказать о поведении E-value лучшей находки и числе находок, использованных для PSSM нельзя. В начале произолшо явное падение числа "плохих" (выделенных желтым) находок, но потом их число вновь стало постепенно расти.
| Итерация | Плохие/Хорошие |
| 9 | 10/490 |
| 10 | 10/490 |
| 11 | 17/483 |
| 12 | 14/496 |
| Таблица 2. Число находок в итерациях 9-12 | |
Стоит отметить, что с каждой следующей итерацией время работы программы несколько увеличивалось, особенно это стало заметно между 11 и 12 итерациями. 11 длилась приблизительно 3 минуты, а вот выполнение 12 составило аж 36 минут. Поэтому она и стала для меня последней.
Данные последней итерации: "хороших" находок - 496, "плохих" - 14, E-value лучшей находки, использованной для PSSM - 5e-119, худшей - 2e-80. E-value худшей находки, использованной для PSSM, и здесь оказался ниже, чем на предыдущих шагах, что согласуется с отмеченной тенденцией. Стабилизации списка мне добиться не удалось. Тем не менее, в целом работа с PSI-BLAST оставляет приятное впечатление